Combining ML and enhanced sampling to unveil the role of dynamics
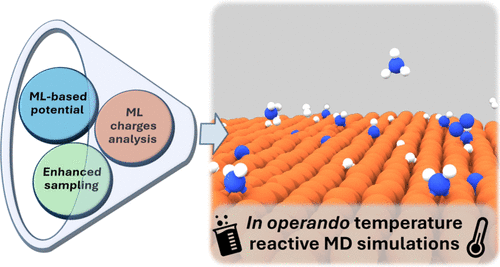
The role of atomic-scale dynamics in heterogeneous catalysis remains poorly understood, especially under operando conditions. In order to study how dynamic effects affect catalytic reactivity, I combined enhanced sampling with machine learning to learn not only the interatomic potentials but also other electronic properties such as the charge transfer, which is an important driving force behind many catalytic processes. We have shown that the dissociative chemisorption of nitrogen on iron, a key step in the Haber–Bosch process, is strongly affected by surface fluctuations at high temperatures, challenging the conventional static picture of catalytic sites (Bonati et al., 2023). Further studies revealed that adsorbed nitrogen species do not poison the catalyst but instead promote dynamic restructuring, which mitigates deactivation (Tripathi et al., 2024). We also applied these methods to study the mechanism of ammonia decomposition (Perego et al., 2024), showing how competition with nitrogen migration within the material affects catalytic efficiency (Purcel et al., 2024), and explaining the promotional role of cobalt alloying in Fe-based catalysts (Perego et al., 2025) within the research network Ammoref. These findings highlight the crucial role of dynamics and open the way for more accurate and predictive models of industrial catalysis.
References
2025
ACS Catal.
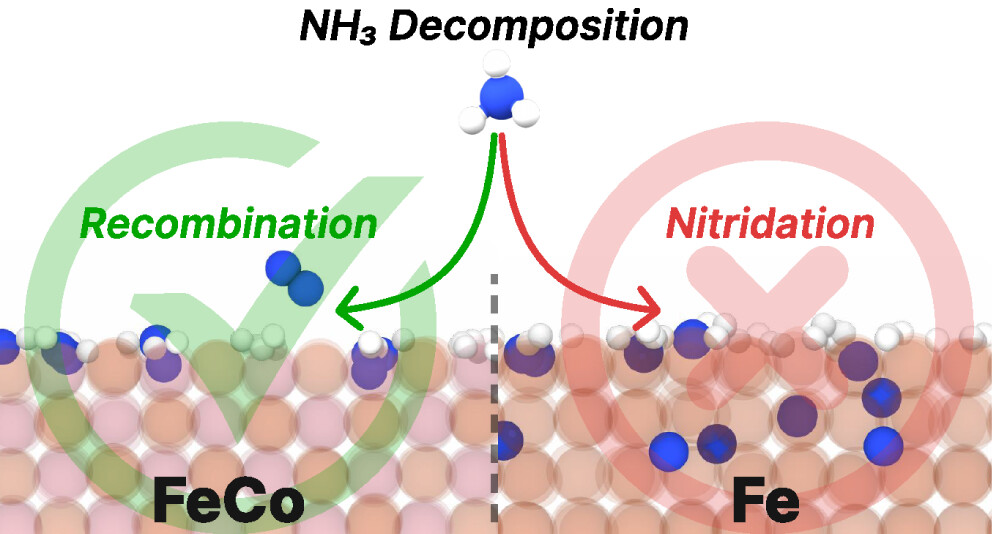
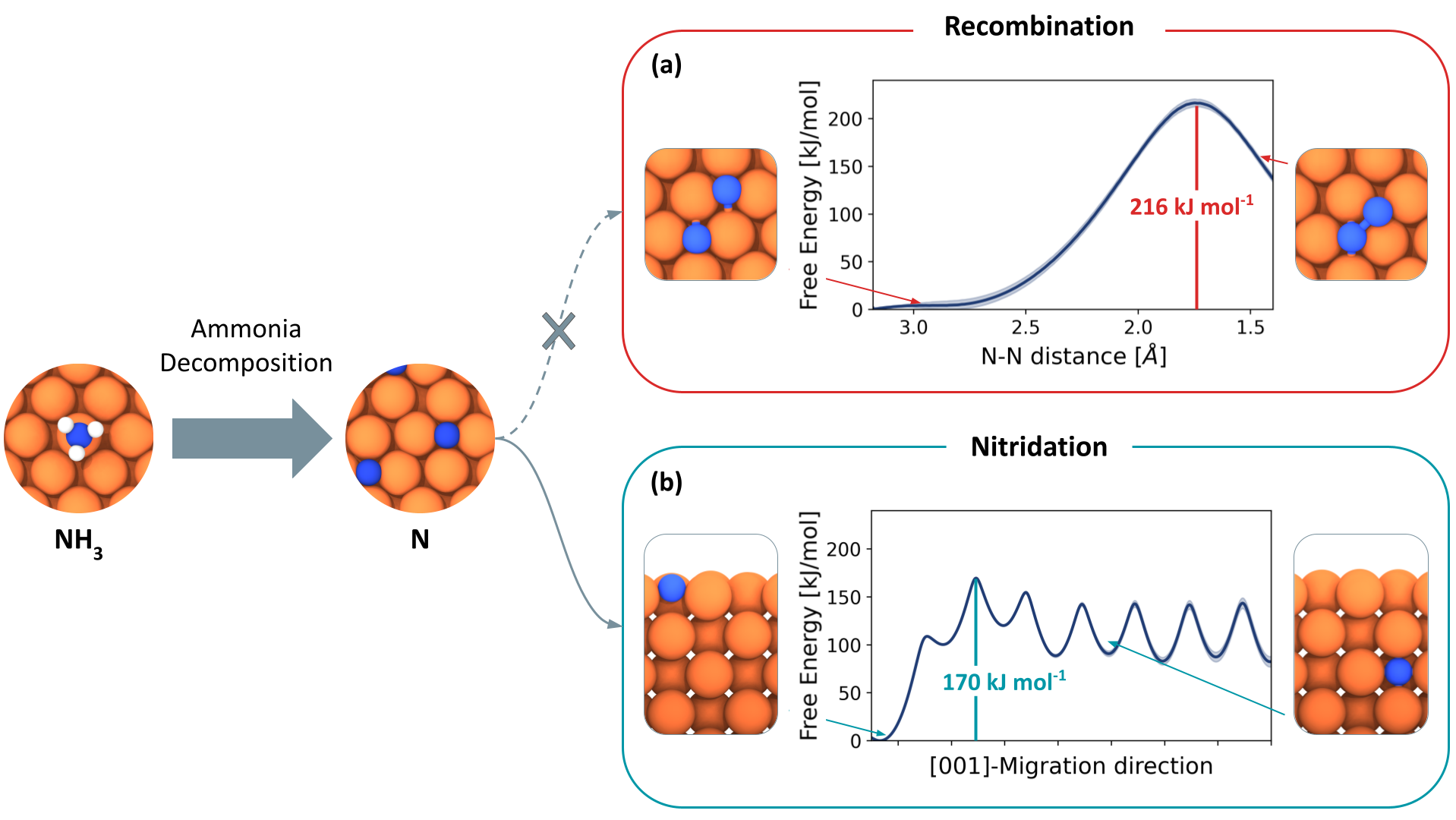
No Time for Nitrides: How Cobalt Alloying Promotes Iron Catalysts for Ammonia Decomposition
Simone Perego, Maximilian Purcel, Yannick Baum, Shilong Chen, Astrid Sophie Müller, Michele Parrinello, Malte Behrens, Martin Muhler, and Luigi Bonati
The increasing demand for hydrogen production has driven interest in ammonia decomposition. Iron-based catalysts, widely used for ammonia synthesis, exhibit suboptimal performance in the reverse pr...
@article{Perego2025NoDecomposition,author={Perego, Simone and Purcel, Maximilian and Baum, Yannick and Chen, Shilong and Müller, Astrid Sophie and Parrinello, Michele and Behrens, Malte and Muhler, Martin and Bonati, Luigi},doi={10.1021/ACSCATAL.5C04795},issn={2155-5435},issue={19},journal={ACS Catalysis},keywords={alloying,ammonia decomposition,machine learning potentials,molecular dynamics,nitridation},month=oct,pages={16690-16702},publisher={American Chemical Society},title={No Time for Nitrides: How Cobalt Alloying Promotes Iron Catalysts for Ammonia Decomposition},volume={15},url={/doi/pdf/10.1021/acscatal.5c04795?ref=article_openPDF},year={2025},}
2024
ACS Catal.
How Poisoning Is Avoided in a Step of Relevance to the Haber-Bosch Catalysis
Shivam Tripathi, Luigi Bonati, Simone Perego, and Michele Parrinello
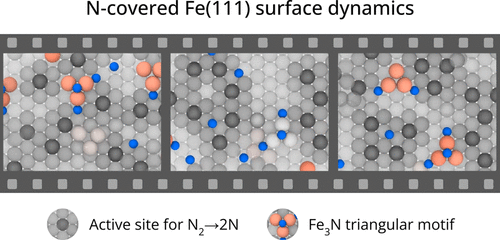
For a catalyst to be efficient and durable, it is crucial that the reaction products do not poison the catalyst. In the case of the Haber-Bosch process, the rate-limiting step is believed to be the decomposition of nitrogen molecules on the Fe(111) surface. This step leads to the production on the surface of atomic nitrogen (N*), which, unless hydrogenated and eventually released as ammonia, remains adsorbed and occupies the active sites. Thus, it is important to ascertain how a high N* coverage affects the nitrogen dissociative chemisorption. To answer this question, we study the properties of the Fe(111) surface at different N* coverage both at room and operando temperature. In the latter regime, we have already found that Fe surface atoms exhibit a high mobility, promoting the formation of adatoms and vacancies, and causing the catalytic centers to acquire a finite lifetime [Bonati et al. Proceedings of the National Academy of Sciences 2023, 120 (50), e2313023120 ]. We discover that the N* coverage reduces but does not eliminate the iron mobility. Remarkably, the N* atoms stabilize triangular surface structures associated with the formation of vacancies, which are a sign of a frustrated drive toward a more stable Fe4N phase. As a consequence, nitrogen atoms tend to cluster, reducing their poisoning effect. At the same time, the reduction in the number of catalytic centers is counteracted by an increase in their lifetime. The combined effect is that the dissociation barrier is not significantly altered in the range of coverages studied. These results bring to light the complex role that dynamics plays in catalytic reactivity under operando conditions.
@article{Tripathi2024HowCatalysis,author={Tripathi, Shivam and Bonati, Luigi and Perego, Simone and Parrinello, Michele},doi={10.1021/ACSCATAL.3C06201},issn={21555435},issue={7},journal={ACS Catalysis },keywords={Haber−Bosch,dissociative chemisorption,iron (111),neural network potential,operando conditions,poisoning,surface dynamics},month=apr,pages={4944-4950},publisher={American Chemical Society},title={How Poisoning Is Avoided in a Step of Relevance to the Haber-Bosch Catalysis},volume={14},url={/doi/pdf/10.1021/acscatal.3c06201?ref=article_openPDF},year={2024},}
ACS Catal.
How Dynamics Changes Ammonia Cracking on Iron Surfaces
Simone Perego, Luigi Bonati, Shivam Tripathi, and Michele Parrinello
Being rich in hydrogen and easy to transport, ammonia is a promising hydrogen carrier. However, a microscopic characterization of the ammonia cracking reaction is still lacking, hindered by extreme operando conditions. Leveraging state-of-the-art molecular dynamics, machine learning potentials, and enhanced sampling methods, we offer an atomistic view of the adsorption, diffusion, and dehydrogenation processes of a single NHx (x = 1, 3) molecule on two representative surfaces at the operando temperature of 700 K. We elucidate the effects of the dynamics on all the steps of decomposition. On the stable (110) surface, we found that the reaction intermediate diffusions are favored over dehydrogenation, with non-negligible effects on the reactivity for one intermediate. The role is even more dramatic on the (111) surface, where the mobility of Fe surface atoms introduces unexplored adsorption sites and significantly alters the dehydrogenation barriers. In both cases, a detailed analysis of reactive events shows that there is never a single transition state, but it is always an ensemble. Notwithstanding, a unified mechanism can be identified by following the charge transfer along the different reaction pathways.
@article{Perego2024HowSurfaces,author={Perego, Simone and Bonati, Luigi and Tripathi, Shivam and Parrinello, Michele},doi={10.1021/ACSCATAL.4C01920},issn={21555435},issue={19},journal={ACS Catalysis},keywords={ammonia decomposition,dynamics,enhanced sampling,green hydrogen,heterogeneous catalysis,machine learning,molecular dynamics,neural network potential},month=oct,pages={14652-14664},publisher={American Chemical Society},title={How Dynamics Changes Ammonia Cracking on Iron Surfaces},volume={14},url={/doi/pdf/10.1021/acscatal.4c01920?ref=article_openPDF},year={2024},}
ACS Catal.
Iron Nitride Formation and Decomposition during Ammonia Decomposition over a Wustite-Based Bulk Iron Catalyst
M. Purcel, S. Berendts, L. Bonati, S. Perego, A. Müller, M. Lerch, M. Parrinello, and M. Muhler
Hydrogen production using renewable sources shows great promise in lowering the dependence on fossil fuels in our current energy system, but challenges persist in its storage and transportation. Thus, ammonia emerges as a viable carrier for H2 due to its favorable properties. NH3 decomposition was studied at atmospheric pressure over a multiply promoted fused FeO-based bulk catalyst optimized for NH3 synthesis by applying transient kinetic experiments, in situ XRD, and Molecular dynamics (MD) simulations. Transient NH3 decomposition and temperature-programmed experiments in a fixed-bed reactor yielded the activation energies of bulk iron nitride formation and decomposition, which occurred in two steps: Fe ⇌ Fe4N ⇌ Fe2N. The decomposition of Fe4N into Fe and N2 was identified as the rate-determining step of NH3 decomposition with essentially the same activation energy amounting to 172 and 173 kJ mol-1, respectively. Fe4N and Fe2N were identified based on the nitrogen mass balance and confirmed as γ′-Fe4N and ϵ-Fe3N1.5 by in situ XRD experiments in NH3. MD simulations showed that the migration of adsorbed N atoms into the bulk of the catalyst is favored over recombinative desorption, influencing N storage and N2 release dynamics. Overall, the findings contribute to a comprehensive understanding of NH3 synthesis catalysts under decomposition conditions, identifying the key descriptor to enhance the performance of Fe-based catalysts.
@article{Purcel2024IronCatalyst,author={Purcel, M. and Berendts, S. and Bonati, L. and Perego, S. and Müller, A. and Lerch, M. and Parrinello, M. and Muhler, M.},doi={10.1021/ACSCATAL.4C04415},issn={21555435},issue={18},journal={ACS Catalysis},keywords={ammonia decomposition,bulk iron catalyst,in situ XRD,machine learning,molecular dynamics simulations,nitridation,transient kinetics},month=sep,pages={13947-13957},publisher={American Chemical Society},title={Iron Nitride Formation and Decomposition during Ammonia Decomposition over a Wustite-Based Bulk Iron Catalyst},volume={14},url={/doi/pdf/10.1021/acscatal.4c04415?ref=article_openPDF},year={2024},}
2023
PNAS
The role of dynamics in heterogeneous catalysis: Surface diffusivity and N2 decomposition on Fe(111)
Luigi Bonati, Daniela Polino, Cristina Pizzolitto, Pierdomenico Biasi, Rene Eckert, Stephan Reitmeier, Robert Schlögl, and Michele Parrinello
Proceedings of the National Academy of Sciences of the United States of America, Dec 2023
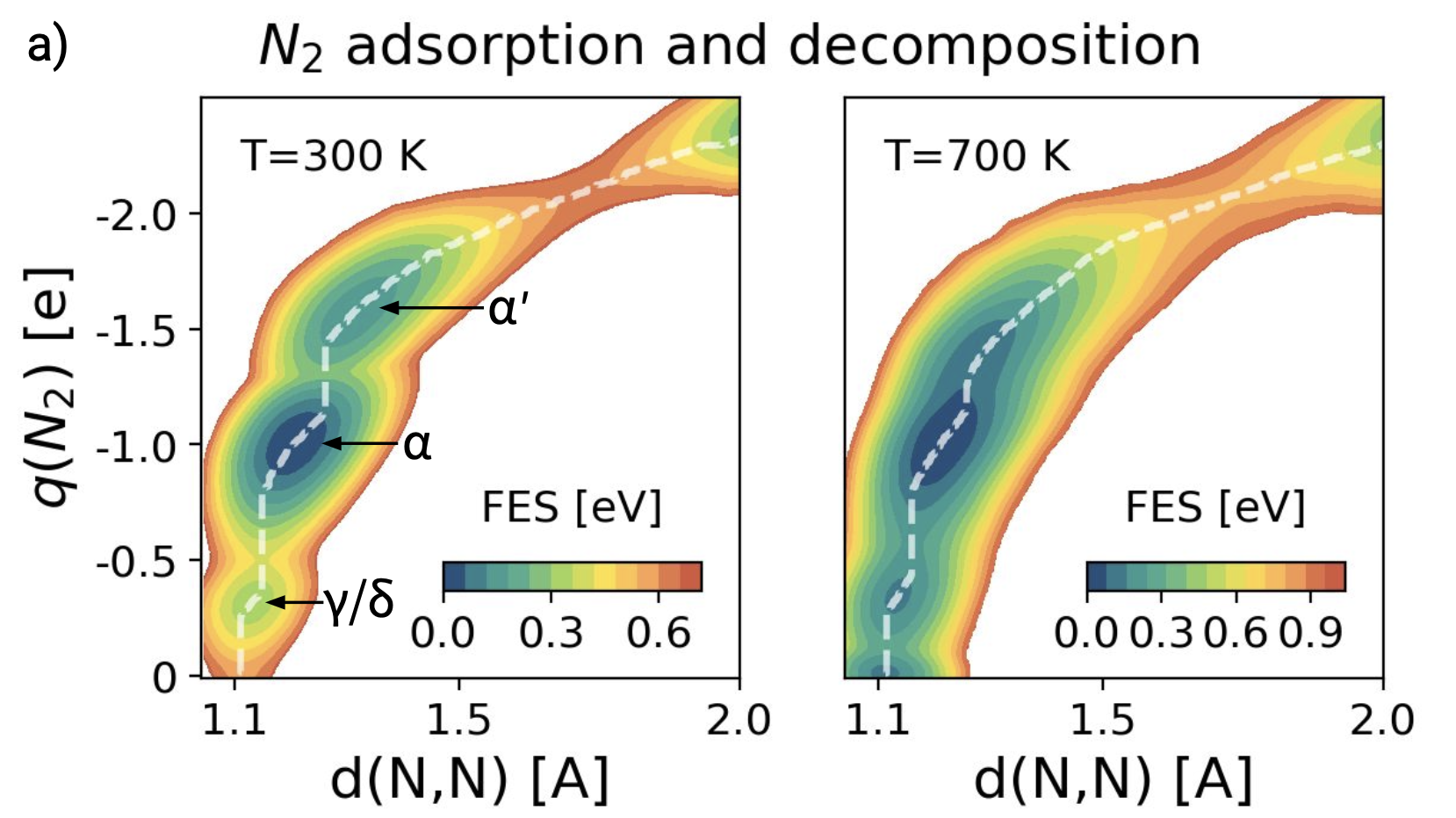
Dynamics has long been recognized to play an important role in heterogeneous catalytic processes. However, until recently, it has been impossible to study their dynamical behavior at industry-relevant temperatures. Using a combination of machine learning potentials and advanced simulation techniques, we investigate the cleavage of the N2 triple bond on the Fe(111) surface. We find that at low temperatures our results agree with the well-established picture. However, if we increase the temperature to reach operando conditions, the surface undergoes a global dynamical change and the step structure of the Fe(111) surface is destabilized. The catalytic sites, traditionally associated with this surface, appear and disappear continuously. Our simulations illuminate the danger of extrapolating low-temperature results to operando conditions and indicate that the catalytic activity can only be inferred from calculations that take dynamics fully into account. More than that, they show that it is the transition to this highly fluctuating interfacial environment that drives the catalytic process.
@article{Bonati2023TheFe111,author={Bonati, Luigi and Polino, Daniela and Pizzolitto, Cristina and Biasi, Pierdomenico and Eckert, Rene and Reitmeier, Stephan and Schlögl, Robert and Parrinello, Michele},doi={10.1073/pnas.231302312},issn={10916490},issue={50},journal={Proceedings of the National Academy of Sciences of the United States of America},keywords={enhanced sampling,heterogeneous catalysis,machine learning,molecular dynamics,nitrogen decomposition},month=dec,pages={e2313023120},pmid={38060558},publisher={National Academy of Sciences},title={The role of dynamics in heterogeneous catalysis: Surface diffusivity and N2 decomposition on Fe(111)},volume={120},url={https://pnas.org/doi/10.1073/pnas.231302312},year={2023},}