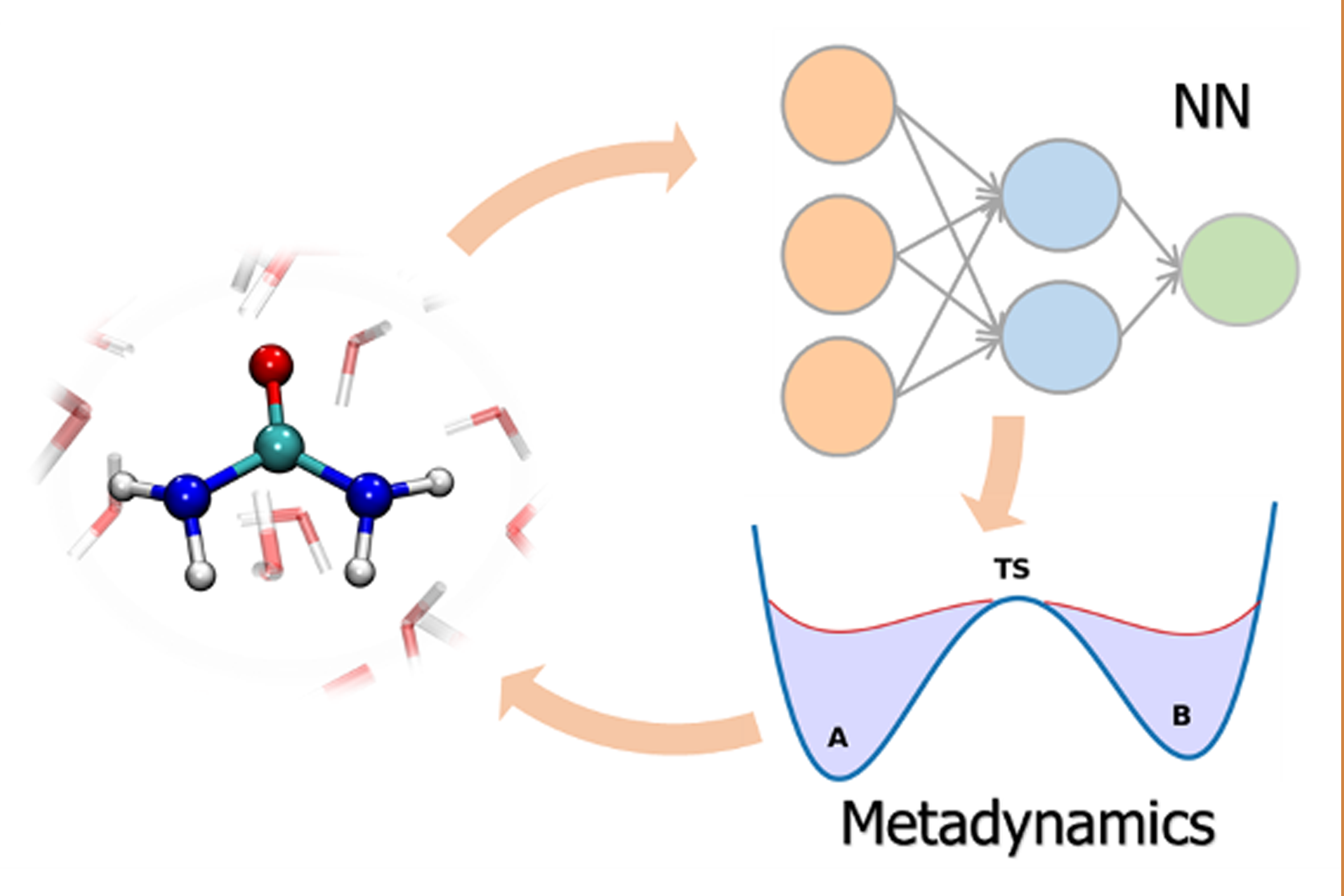
Using enhanced sampling to build ML potentials for rare events. The construction of ML interatomic potentials for phase transitions and chemical reactions is challenging due to the difficulty of including all relevant configurations in the training set. By integrating enhanced sampling techniques into active learning strategies we are able obtain reliable and robust machine learning potentials. This enables ab initio-quality simulations of rare events which would otherwise be prohibitively expensive, ranging from crystallization (Bonati & Parrinello, 2018) to phase diagrams (Niu et al., 2020) and from chemical reactions in solvent (Yang et al., 2022) to heterogeneous catalysis (Bonati et al., 2023) and to phase-change materials (Kheir et al., 2024).
Data-efficient active learning. To make the machine learning potentials routinely applicable and to model processes in more realistic conditions and with higher levels of electronic theory, it is essential to have data-efficient techniques. To this end, I have devised a framework that integrates advanced sampling with Gaussian processes and graph neural networks to construct reactive potentials in a highly efficient manner (Perego & Bonati, 2024). This data-efficient active learning (DEAL) scheme enables an ab initio-quality discovery of transition paths and ensures uniform accuracy along them, with a 20-fold increase in data-efficiency with respect to previous approaches.
Left: gaussian-process based enhanced sampling exploration of reaction pathways. Right: data-efficient active learning selection.
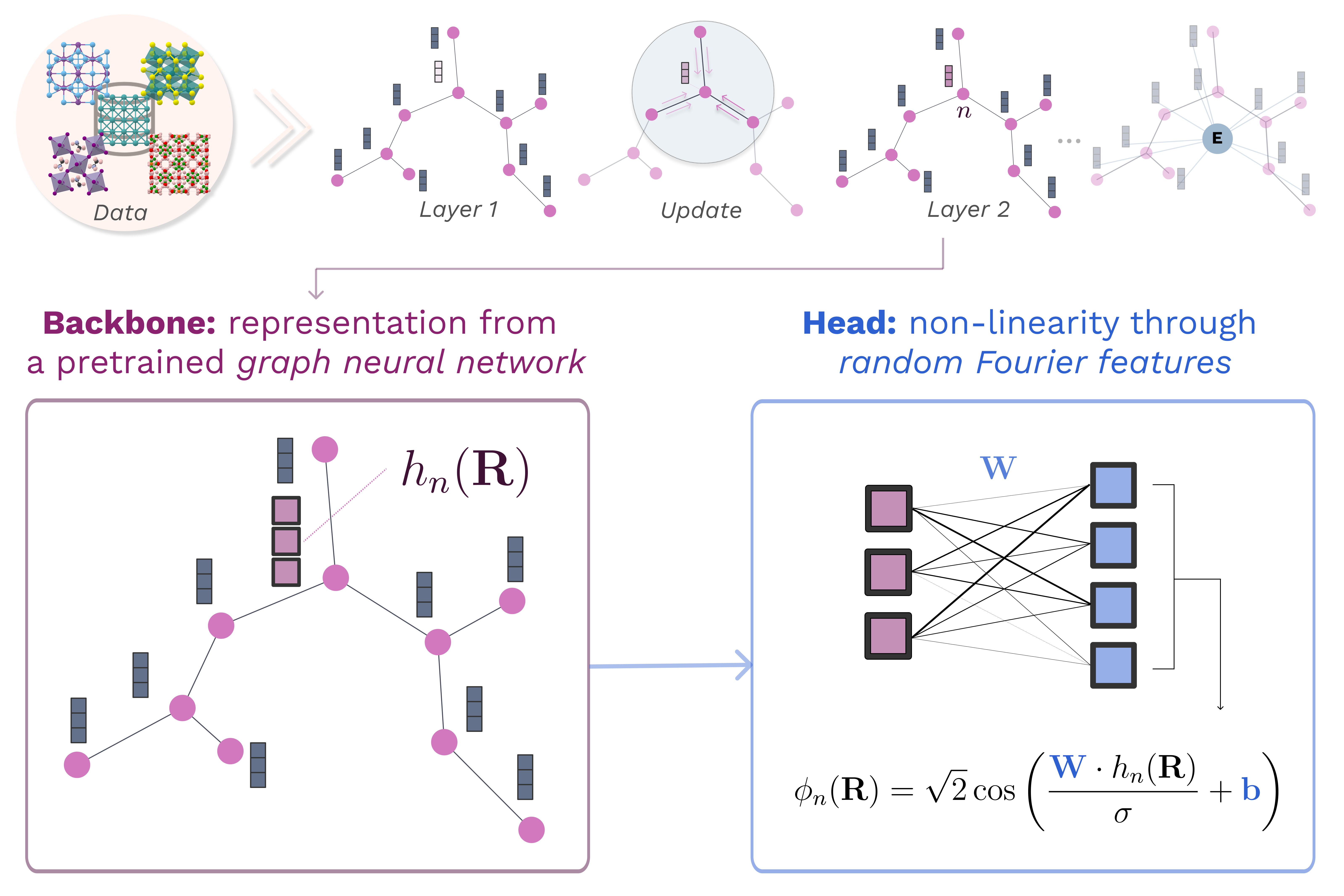
Transfer learning for atomistic simulations. Furthermore, we are also developing transfer learning approaches to extract the representation learned from graph neural networks trained on large datasets and transfer them to new systems via kernel methods (Falk et al., 2023). In particular, we combined them with random Fourier features, a large-scale kernel approximation. (Novelli et al., 2025). This also provides a closed-form fine-tuning strategy for general-purpose potentials such as MACE-MP0, enabling fast and accurate adaptation to new systems or levels of quantum mechanical theory with minimal hyperparameter tuning. This provides a data-efficient framework not only for energy/force predictions but also for stable and accurate MD simulations using just a few tens of training data.
Training machine learning interatomic potentials that are both computationally and data-efficient is a key challenge for enabling their routine use in atomistic simulations. To this effect, we introduce , a scalable and lightweight transfer learning framework that extracts atomic descriptors from pre-trained graph neural networks and transfers them to new systems using random Fourier features — an efficient and scalable approximation of kernel methods. It also provides a closed-form fine-tuning strategy for general-purpose potentials such as MACE-MP0, enabling fast and accurate adaptation to new systems or levels of quantum mechanical theory with minimal hyperparameter tuning. On a benchmark dataset of 27 transition metals, outperforms optimized kernel-based methods in both training time and accuracy, reducing model training from tens of hours to minutes on a single GPU. We further demonstrate the framework’s strong data-efficiency by training stable and accurate potentials for bulk water and the Pt(111)/water interface using just tens of training structures. Our open-source implementation ( https://franken.readthedocs.io ) offers a fast and practical solution for training potentials and deploying them for molecular dynamics simulations across diverse systems.
@article{Novelli2025FastPotentials,author={Novelli, Pietro and Meanti, Giacomo and Buigues, Pedro J. and Rosasco, Lorenzo and Parrinello, Michele and Pontil, Massimiliano and Bonati, Luigi},doi={10.1038/s41524-025-01779-z},issn={2057-3960},issue={1},journal={npj Computational Materials},keywords={Condensed,Physical chemistry,Theoretical chemistry,matter physics},month=sep,pages={293},publisher={Nature Publishing Group},title={Fast and Fourier features for transfer learning of interatomic potentials},volume={11},url={https://www.nature.com/articles/s41524-025-01779-z},year={2025},}
2024
npj Comput. Mater.
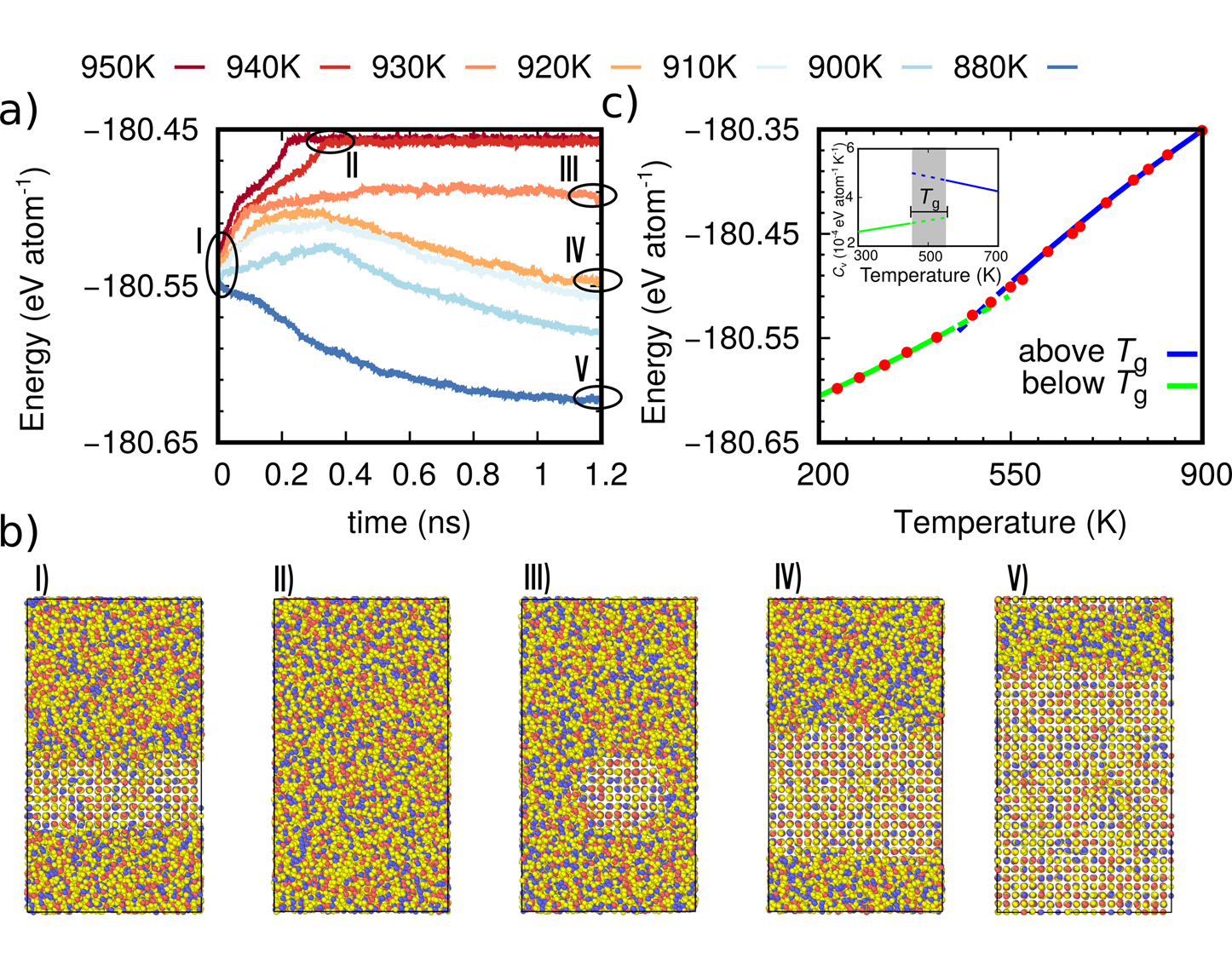
Unraveling the crystallization kinetics of the Ge2Sb2Te5 phase change compound with a machine-learned interatomic potential
Omar Abou El Kheir, Luigi Bonati, Michele Parrinello, and Marco Bernasconi
The phase change compound Ge2Sb2Te5 (GST225) is exploited in advanced non-volatile electronic memories and in neuromorphic devices which both rely on a fast and reversible transition between the crystalline and amorphous phases induced by Joule heating. The crystallization kinetics of GST225 is a key functional feature for the operation of these devices. We report here on the development of a machine-learned interatomic potential for GST225 that allowed us to perform large scale molecular dynamics simulations (over 10,000 atoms for over 100 ns) to uncover the details of the crystallization kinetics in a wide range of temperatures of interest for the programming of the devices. The potential is obtained by fitting with a deep neural network (NN) scheme a large quantum-mechanical database generated within density functional theory. The availability of a highly efficient and yet highly accurate NN potential opens the possibility to simulate phase change materials at the length and time scales of the real devices.
@article{AbouElKheir2024UnravelingPotential,author={Kheir, Omar Abou El and Bonati, Luigi and Parrinello, Michele and Bernasconi, Marco},doi={10.1038/S41524-024-01217-6},issn={20573960},issue={1},journal={npj Computational Materials},keywords={Atomistic models,Electronic devices,Structure of solids and liquids},month=dec,pages={1-12},publisher={Nature Research},title={Unraveling the crystallization kinetics of the Ge2Sb2Te5 phase change compound with a machine-learned interatomic potential},volume={10},url={https://www.nature.com/articles/s41524-024-01217-6},year={2024},}
npj Comput. Mater.
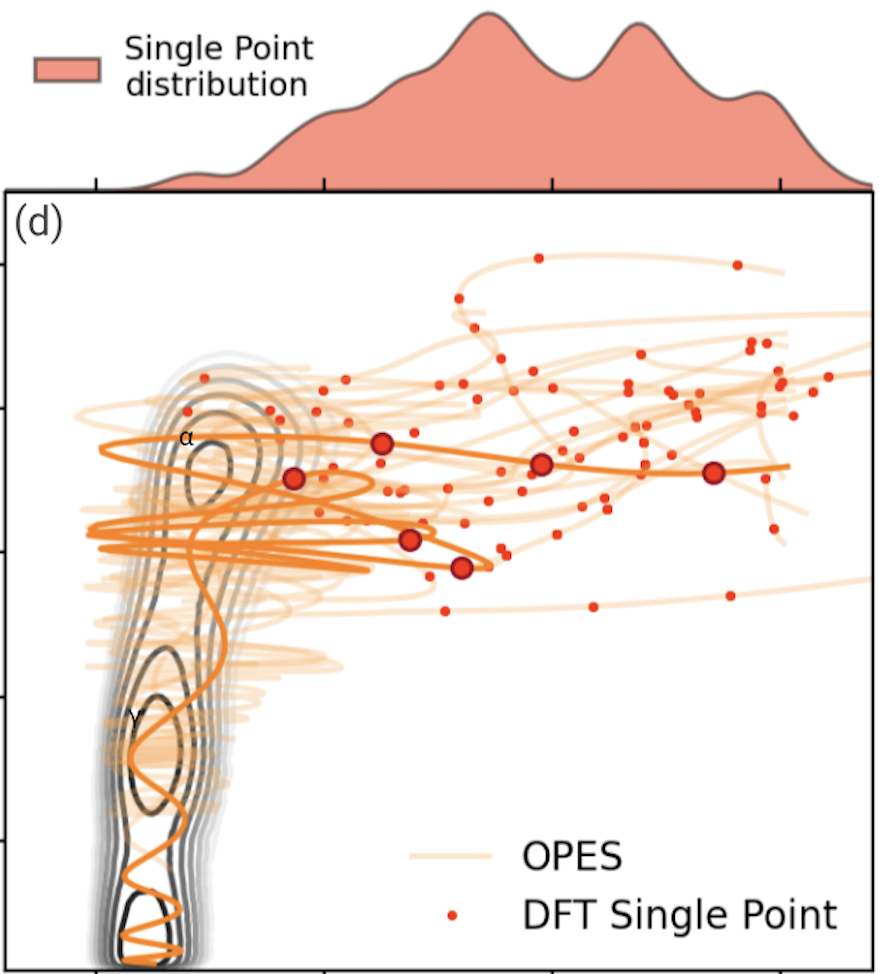
Data efficient machine learning potentials for modeling catalytic reactivity via active learning and enhanced sampling
Simulating catalytic reactivity under operative conditions poses a significant challenge due to the dynamic nature of the catalysts and the high computational cost of electronic structure calculations. Machine learning potentials offer a promising avenue to simulate dynamics at a fraction of the cost, but they require datasets containing all relevant configurations, particularly reactive ones. Here, we present a scheme to construct reactive potentials in a data-efficient manner. This is achieved by combining enhanced sampling methods first with Gaussian processes to discover transition paths and then with graph neural networks to obtain a uniformly accurate description. The necessary configurations are extracted via a Data-Efficient Active Learning (DEAL) procedure based on local environment uncertainty. We validated our approach by studying several reactions related to the decomposition of ammonia on iron-cobalt alloy catalysts. Our scheme proved to be efficient, requiring only 1000 DFT calculations per reaction, and robust, sampling reactive configurations from the different accessible pathways. Using this potential, we calculated free energy profiles and characterized reaction mechanisms, showing the ability to provide microscopic insights into complex processes under dynamic conditions.
@article{Perego2024DataSampling,author={Perego, Simone and Bonati, Luigi},doi={10.1038/S41524-024-01481-6},issn={20573960},issue={1},journal={npj Computational Materials},keywords={Atomistic models,Computational methods,Heterogeneous catalysis,Theoretical chemistry},month=dec,pages={1-13},publisher={Nature Research},title={Data efficient machine learning potentials for modeling catalytic reactivity via active learning and enhanced sampling},volume={10},url={https://www.nature.com/articles/s41524-024-01481-6},year={2024},}
2023
PNAS
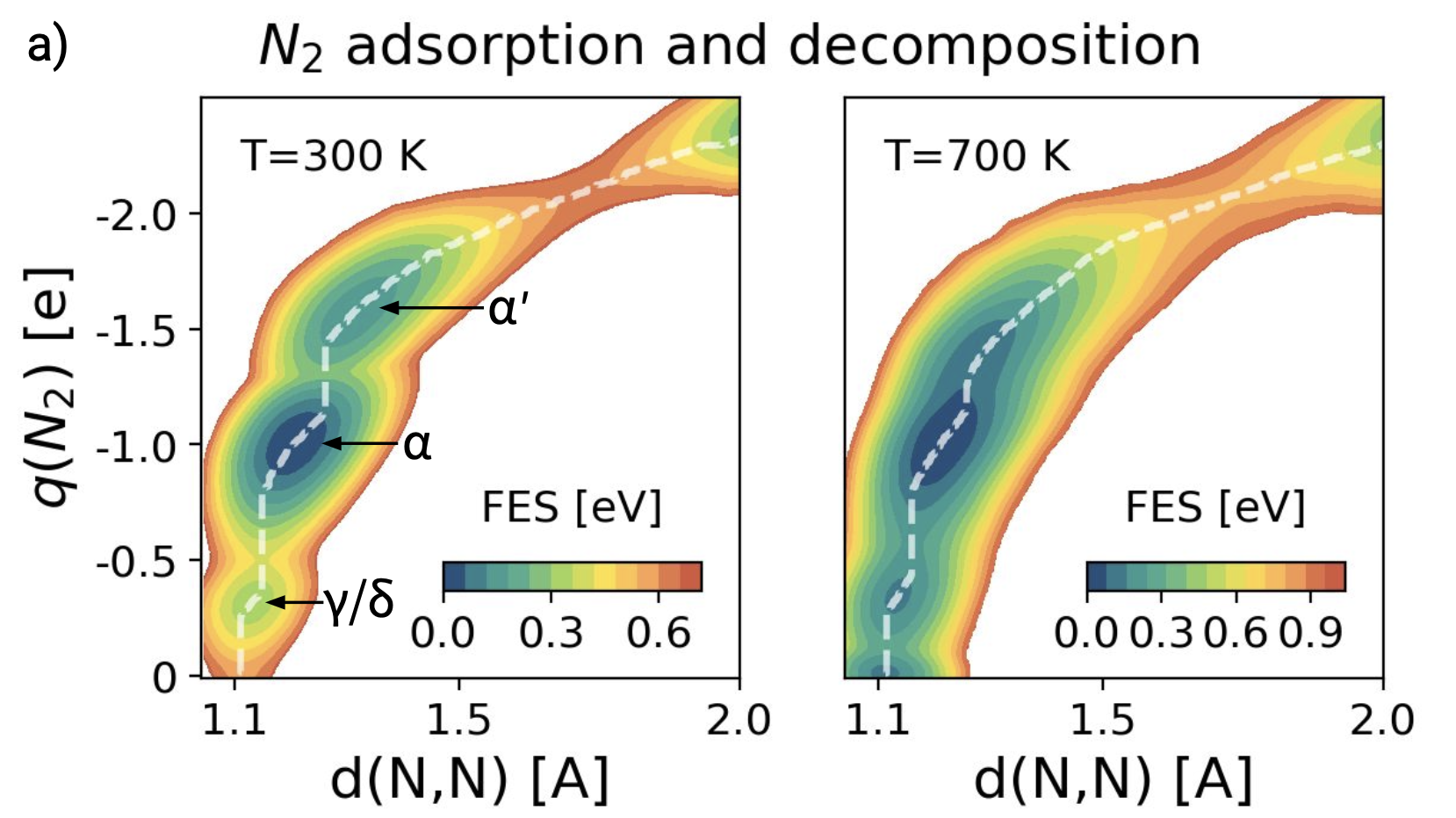
The role of dynamics in heterogeneous catalysis: Surface diffusivity and N2 decomposition on Fe(111)
Luigi Bonati, Daniela Polino, Cristina Pizzolitto, Pierdomenico Biasi, Rene Eckert, Stephan Reitmeier, Robert Schlögl, and Michele Parrinello
Proceedings of the National Academy of Sciences of the United States of America, Dec 2023
Dynamics has long been recognized to play an important role in heterogeneous catalytic processes. However, until recently, it has been impossible to study their dynamical behavior at industry-relevant temperatures. Using a combination of machine learning potentials and advanced simulation techniques, we investigate the cleavage of the N2 triple bond on the Fe(111) surface. We find that at low temperatures our results agree with the well-established picture. However, if we increase the temperature to reach operando conditions, the surface undergoes a global dynamical change and the step structure of the Fe(111) surface is destabilized. The catalytic sites, traditionally associated with this surface, appear and disappear continuously. Our simulations illuminate the danger of extrapolating low-temperature results to operando conditions and indicate that the catalytic activity can only be inferred from calculations that take dynamics fully into account. More than that, they show that it is the transition to this highly fluctuating interfacial environment that drives the catalytic process.
@article{Bonati2023TheFe111,author={Bonati, Luigi and Polino, Daniela and Pizzolitto, Cristina and Biasi, Pierdomenico and Eckert, Rene and Reitmeier, Stephan and Schlögl, Robert and Parrinello, Michele},doi={10.1073/pnas.231302312},issn={10916490},issue={50},journal={Proceedings of the National Academy of Sciences of the United States of America},keywords={enhanced sampling,heterogeneous catalysis,machine learning,molecular dynamics,nitrogen decomposition},month=dec,pages={e2313023120},pmid={38060558},publisher={National Academy of Sciences},title={The role of dynamics in heterogeneous catalysis: Surface diffusivity and N2 decomposition on Fe(111)},volume={120},url={https://pnas.org/doi/10.1073/pnas.231302312},year={2023},}
Neurips
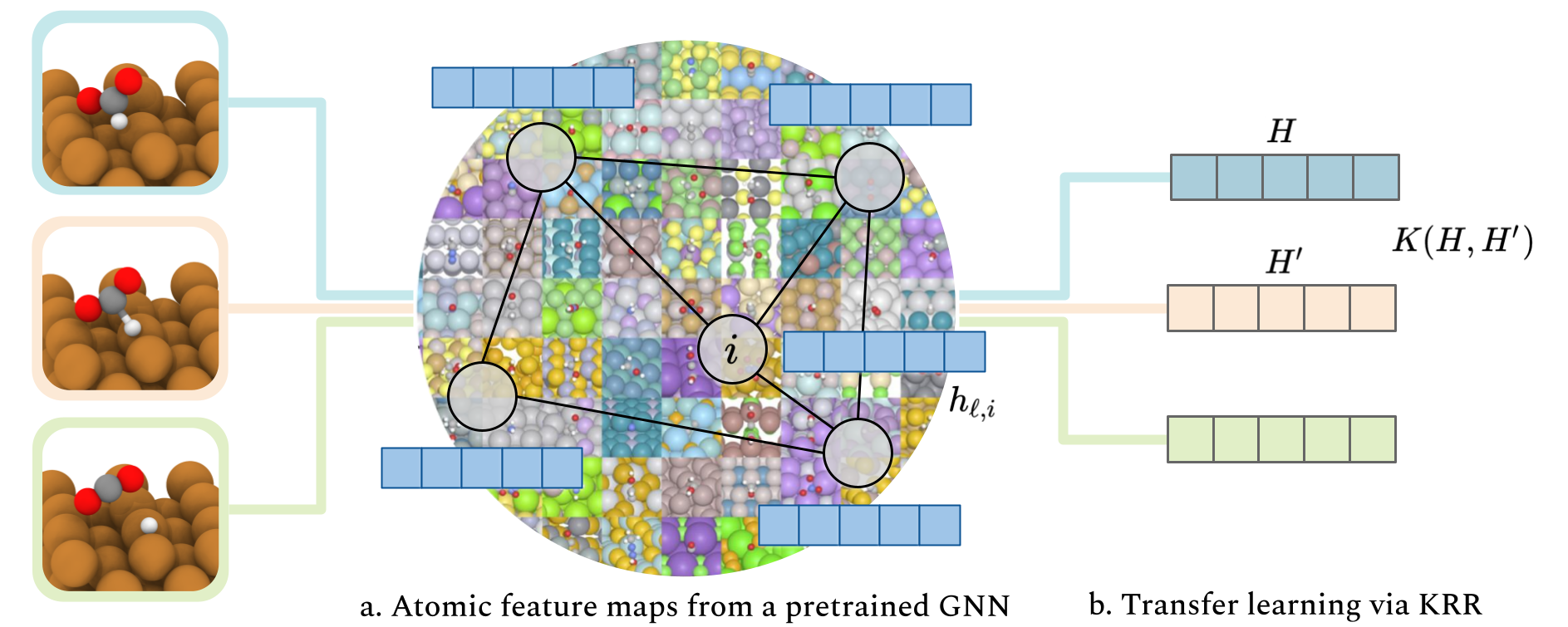
Transfer learning for atomistic simulations using GNNs and kernel mean embeddings
John Falk, Luigi Bonati, Pietro Novelli, Michele Parrinello, and Massimiliano Pontil
Advances in Neural Information Processing Systems, 2023
@article{Falk2023Transfer,title={Transfer learning for atomistic simulations using GNNs and kernel mean embeddings},author={Falk, John and Bonati, Luigi and Novelli, Pietro and Parrinello, Michele and Pontil, Massimiliano},journal={Advances in Neural Information Processing Systems},volume={36},pages={29783--29797},year={2023},}
2022
Catal. Today
Using metadynamics to build neural network potentials for reactive events: the case of urea decomposition in water
Manyi Yang, Luigi Bonati, Daniela Polino, and Michele Parrinello
The study of chemical reactions in aqueous media is very important for its implications in several fields of science, from biology to industrial processes. However, modeling these reactions is difficult when water directly participates in the reaction, since it requires a fully quantum mechanical description of the system. Ab-initio molecular dynamics is the ideal candidate to shed light on these processes. However, its scope is limited by a high computational cost. A popular alternative is to perform molecular dynamics simulations powered by machine learning potentials, trained on an extensive set of quantum mechanical calculations. Doing so reliably for reactive processes is difficult because it requires including very many intermediate and transition state configurations. In this study we used an active learning procedure accelerated by enhanced sampling to harvest such structures and to build a neural-network potential to study the urea decomposition process in water. This allowed us to obtain the free energy profiles of this important reaction in a wide range of temperatures, to discover several novel metastable states, and improve the accuracy of the kinetic rates calculations. Furthermore, we found that the formation of the zwitterionic intermediate has the same probability of occurring via an acidic or a basic pathway, which could be the cause of the insensitivity of reaction rates to the solution pH.
@article{Yang2022UsingWater,author={Yang, Manyi and Bonati, Luigi and Polino, Daniela and Parrinello, Michele},doi={10.1016/J.CATTOD.2021.03.018},issn={0920-5861},journal={Catalysis Today},keywords={Free energy surface,Kinetic rates,Metadynamics,Neural network potentials,Urea decomposition},month=mar,pages={143-149},publisher={Elsevier},title={Using metadynamics to build neural network potentials for reactive events: the case of urea decomposition in water},volume={387},url={https://www.sciencedirect.com/science/article/pii/S092058612100136X},year={2022},}
2020
Nat. Commun.
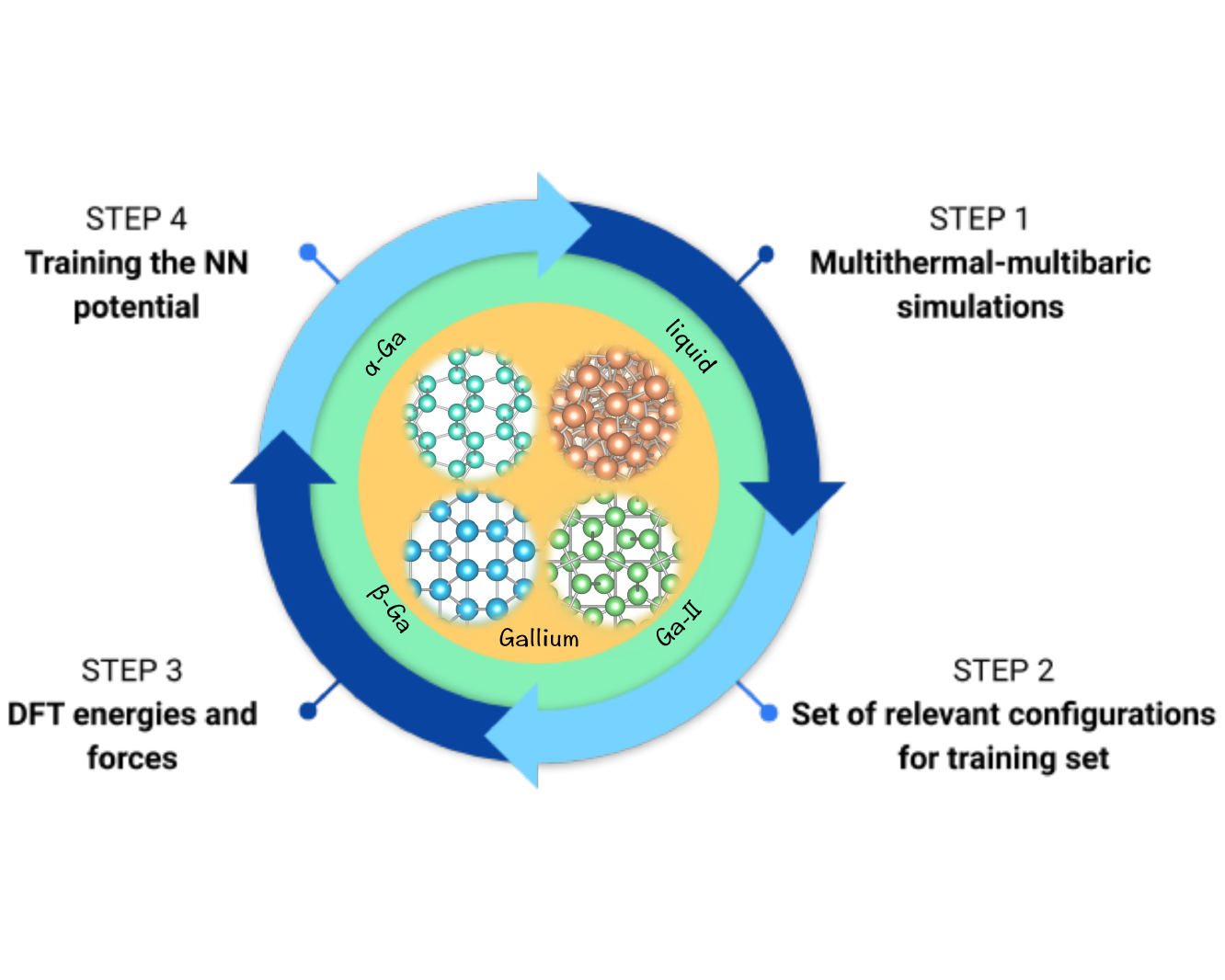
Ab initio phase diagram and nucleation of gallium
Haiyang Niu, Luigi Bonati, Pablo M. Piaggi, and Michele Parrinello
Elemental gallium possesses several intriguing properties, such as a low melting point, a density anomaly and an electronic structure in which covalent and metallic features coexist. In order to simulate this complex system, we construct an ab initio quality interaction potential by training a neural network on a set of density functional theory calculations performed on configurations generated in multithermal–multibaric simulations. Here we show that the relative equilibrium between liquid gallium, α-Ga, β-Ga, and Ga-II is well described. The resulting phase diagram is in agreement with the experimental findings. The local structure of liquid gallium and its nucleation into α-Ga and β-Ga are studied. We find that the formation of metastable β-Ga is kinetically favored over the thermodinamically stable α-Ga. Finally, we provide insight into the experimental observations of extreme undercooling of liquid Ga.
@article{Niu2020AbGallium,author={Niu, Haiyang and Bonati, Luigi and Piaggi, Pablo M. and Parrinello, Michele},doi={10.1038/s41467-020-16372-9},issn={20411723},issue={1},journal={Nature Communications},pages={1-9},pmid={32461573},publisher={Springer US},title={Ab initio phase diagram and nucleation of gallium},volume={11},url={http://dx.doi.org/10.1038/s41467-020-16372-9},year={2020},}
2018
PRL
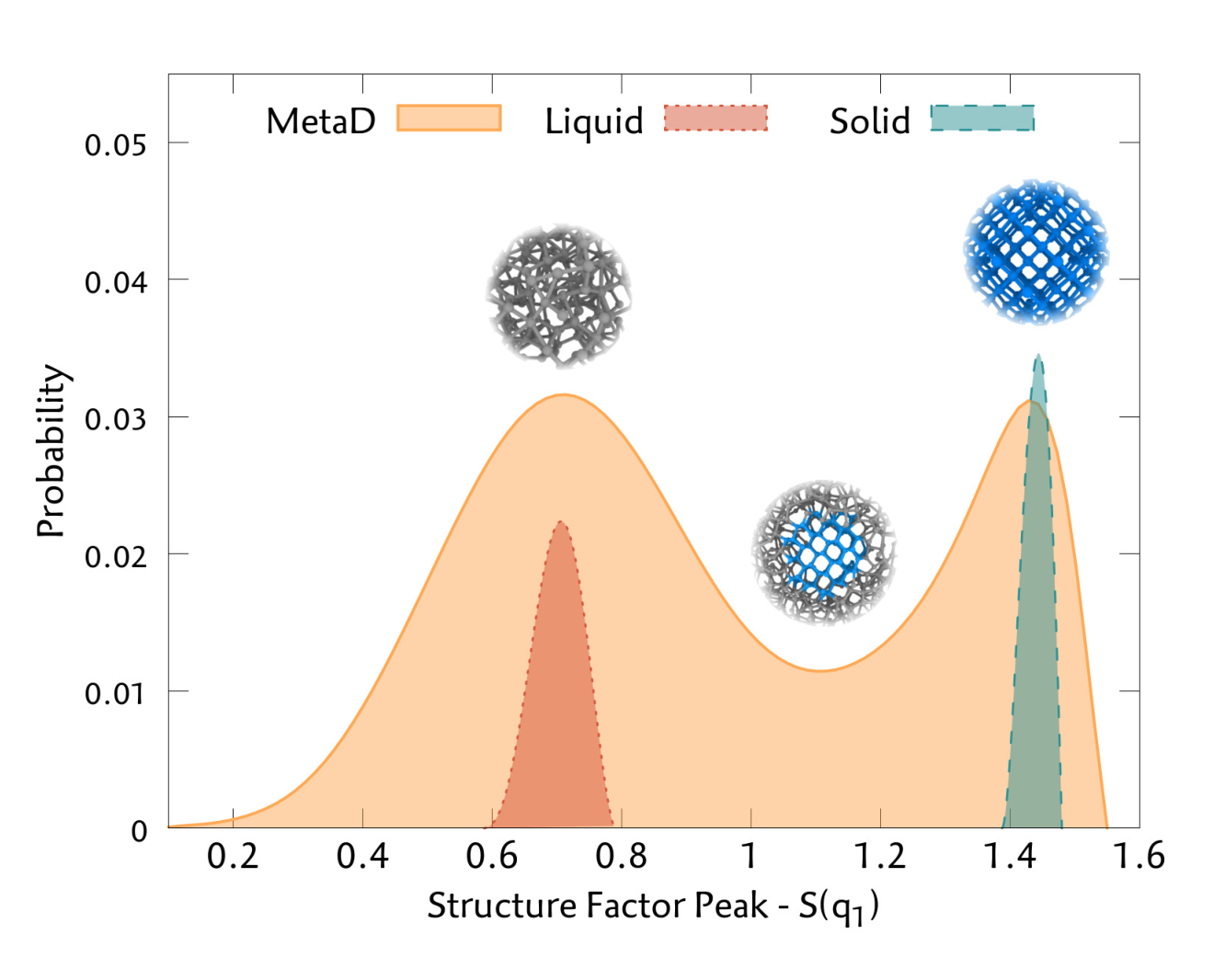
Silicon Liquid Structure and Crystal Nucleation from Ab Initio Deep Metadynamics
Studying the crystallization process of silicon is a challenging task since empirical potentials are not able to reproduce well the properties of both a semiconducting solid and metallic liquid. On the other hand, nucleation is a rare event that occurs in much longer timescales than those achievable by ab initio molecular dynamics. To address this problem, we train a deep neural network potential based on a set of data generated by metadynamics simulations using a classical potential. We show how this is an effective way to collect all the relevant data for the process of interest. In order to efficiently drive the crystallization process, we introduce a new collective variable based on the Debye structure factor. We are able to encode the long-range order information in a local variable which is better suited to describe the nucleation dynamics. The reference energies are then calculated using the strongly constrained and appropriately normed (SCAN) exchange-correlation functional, which is able to get a better description of the bonding complexity of the Si phase diagram. Finally, we recover the free energy surface with a density functional theory accuracy, and we compute the thermodynamics properties near the melting point, obtaining a good agreement with experimental data. In addition, we study the early stages of the crystallization process, unveiling features of the nucleation mechanism.
@article{Bonati2018SiliconMetadynamics,author={Bonati, Luigi and Parrinello, Michele},doi={10.1103/PhysRevLett.121.265701},issn={0031-9007},issue={26},journal={Physical Review Letters},month=dec,pages={265701},pmid={30636123},title={Silicon Liquid Structure and Crystal Nucleation from <i>Ab Initio</i> Deep Metadynamics},volume={121},url={https://link.aps.org/doi/10.1103/PhysRevLett.121.265701},year={2018},}





 Transfer learning for atomistic simulations using GNNs and kernel mean embeddingsAdvances in Neural Information Processing Systems, 2023
Transfer learning for atomistic simulations using GNNs and kernel mean embeddingsAdvances in Neural Information Processing Systems, 2023