publications
See my Google Scholar profile for the most updated list.
2025
- npj Comput. Mater.
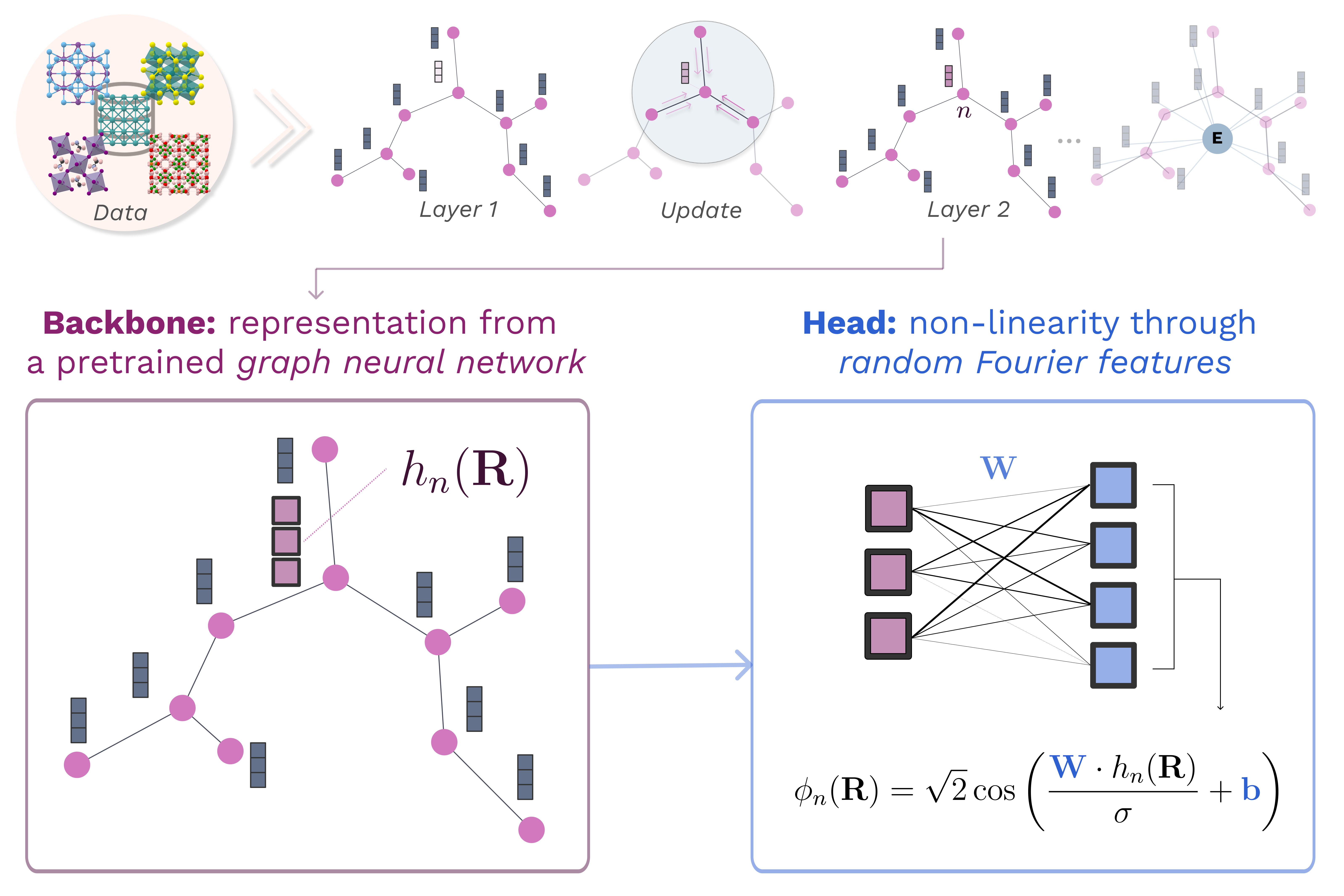 Fast and Fourier features for transfer learning of interatomic potentialsPietro Novelli, Giacomo Meanti, Pedro J. Buigues, Lorenzo Rosasco, Michele Parrinello, Massimiliano Pontil, and Luigi Bonatinpj Computational Materials, Sep 2025
Fast and Fourier features for transfer learning of interatomic potentialsPietro Novelli, Giacomo Meanti, Pedro J. Buigues, Lorenzo Rosasco, Michele Parrinello, Massimiliano Pontil, and Luigi Bonatinpj Computational Materials, Sep 2025Training machine learning interatomic potentials that are both computationally and data-efficient is a key challenge for enabling their routine use in atomistic simulations. To this effect, we introduce , a scalable and lightweight transfer learning framework that extracts atomic descriptors from pre-trained graph neural networks and transfers them to new systems using random Fourier features — an efficient and scalable approximation of kernel methods. It also provides a closed-form fine-tuning strategy for general-purpose potentials such as MACE-MP0, enabling fast and accurate adaptation to new systems or levels of quantum mechanical theory with minimal hyperparameter tuning. On a benchmark dataset of 27 transition metals, outperforms optimized kernel-based methods in both training time and accuracy, reducing model training from tens of hours to minutes on a single GPU. We further demonstrate the framework’s strong data-efficiency by training stable and accurate potentials for bulk water and the Pt(111)/water interface using just tens of training structures. Our open-source implementation (
https://franken.readthedocs.io ) offers a fast and practical solution for training potentials and deploying them for molecular dynamics simulations across diverse systems.@article{Novelli2025FastPotentials, author = {Novelli, Pietro and Meanti, Giacomo and Buigues, Pedro J. and Rosasco, Lorenzo and Parrinello, Michele and Pontil, Massimiliano and Bonati, Luigi}, doi = {10.1038/s41524-025-01779-z}, issn = {2057-3960}, issue = {1}, journal = {npj Computational Materials}, keywords = {Condensed,Physical chemistry,Theoretical chemistry,matter physics}, month = sep, pages = {293}, publisher = {Nature Publishing Group}, title = {Fast and Fourier features for transfer learning of interatomic potentials}, volume = {11}, url = {https://www.nature.com/articles/s41524-025-01779-z}, year = {2025}, } - PNAS
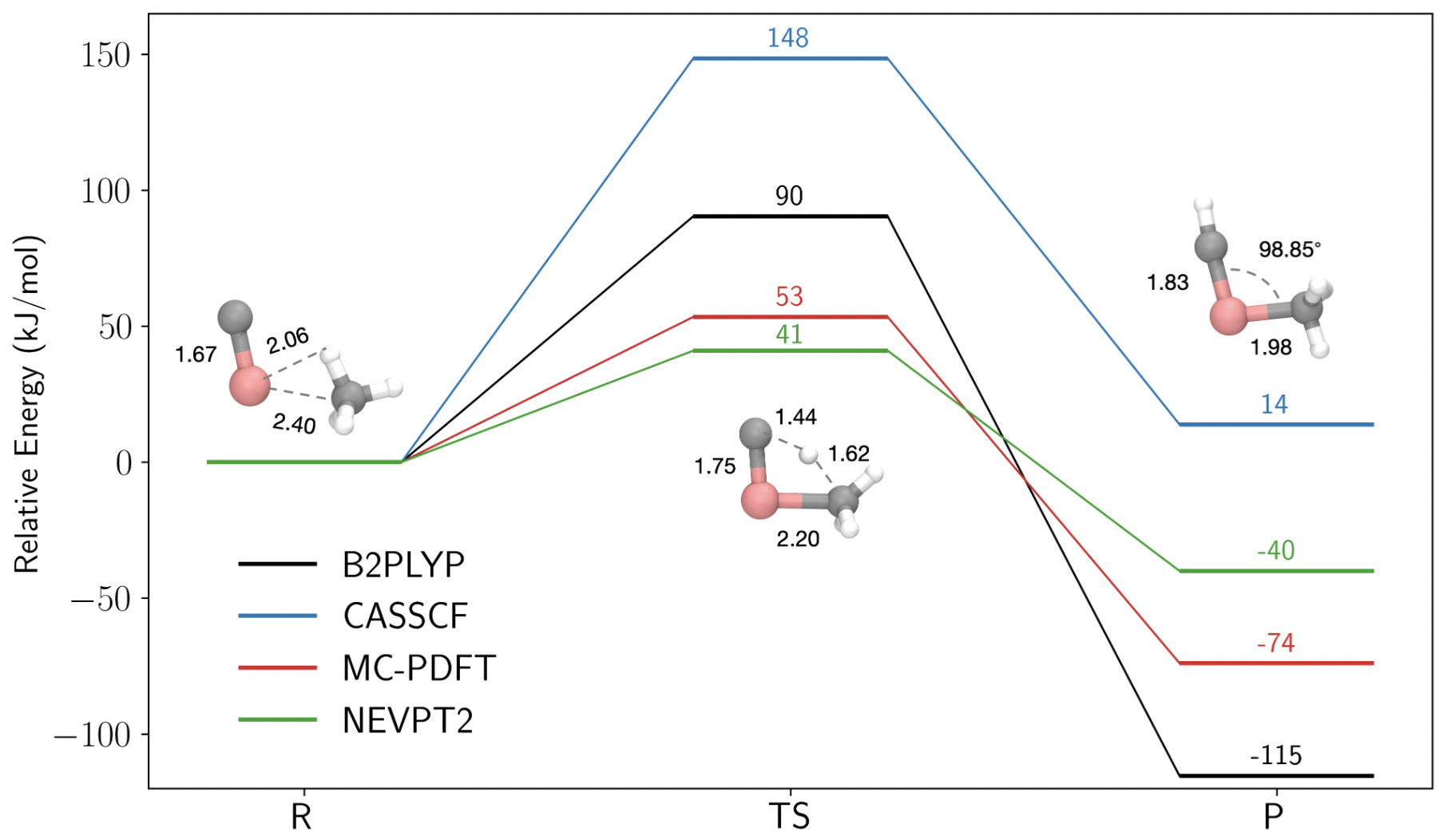 Weighted active space protocol for multireference machine-learned potentialsAniruddha Seal, Simone Perego, Matthew R. Hennefarth, Umberto Raucci, Luigi Bonati, Andrew L. Ferguson, Michele Parrinello, and Laura GagliardiProceedings of the National Academy of Sciences, Sep 2025
Weighted active space protocol for multireference machine-learned potentialsAniruddha Seal, Simone Perego, Matthew R. Hennefarth, Umberto Raucci, Luigi Bonati, Andrew L. Ferguson, Michele Parrinello, and Laura GagliardiProceedings of the National Academy of Sciences, Sep 2025Multireference methods such as multiconfiguration pair-density functional theory accurately capture electronic correlation in systems with strong multiconfigurational character, but their cost precludes direct use in molecular dynamics. Combining these methods with machine-learned interatomic potentials (MLPs) can extend their reach. However, the sensitivity of multireference calculations to the choice of the active space complicates the consistent evaluation of energies and gradients across structurally diverse nuclear configurations. To overcome this limitation, we introduce the weighted active space protocol (WASP), a systematic approach to assign a consistent active space for a given system across uncorrelated configurations. By integrating WASP with MLPs and enhanced sampling techniques, we propose a data-efficient active learning cycle that enables the training of an MLP on multireference data. We demonstrated the approach on the TiC + -catalyzed C–H activation of methane, a reaction that poses challenges for Kohn–Sham density functional theory due to its significant multireference character. This framework enables accurate and efficient modeling of catalytic dynamics, establishing a paradigm for simulating complex reactive processes beyond the limits of conventional electronic-structure methods.
@article{Seal2025WeightedPotentials, author = {Seal, Aniruddha and Perego, Simone and Hennefarth, Matthew R. and Raucci, Umberto and Bonati, Luigi and Ferguson, Andrew L. and Parrinello, Michele and Gagliardi, Laura}, doi = {10.1073/pnas.2513693122}, issn = {0027-8424}, issue = {38}, journal = {Proceedings of the National Academy of Sciences}, keywords = {enhanced sampling,machine-learned potentials,molecular dynamics,strong correlation}, month = sep, pages = {e2513693122}, pmid = {40953275}, publisher = {National Academy of Sciences}, title = {Weighted active space protocol for multireference machine-learned potentials}, volume = {122}, url = {https://pnas.org/doi/10.1073/pnas.2513693122}, year = {2025}, } - Chem. Rev.
 Enhanced Sampling in the Age of Machine Learning: Algorithms and ApplicationsKai Zhu, Enrico Trizio, Jintu Zhang, Renling Hu, Linlong Jiang, Tingjun Hou, and Luigi BonatiChemical Reviews, Oct 2025
Enhanced Sampling in the Age of Machine Learning: Algorithms and ApplicationsKai Zhu, Enrico Trizio, Jintu Zhang, Renling Hu, Linlong Jiang, Tingjun Hou, and Luigi BonatiChemical Reviews, Oct 2025Molecular dynamics simulations hold great promise for providing insight into the microscopic behavior of complex molecular systems. However, their effectiveness is often constrained by long timescales associated with rare events. Enhanced sampling methods have been developed to address these challenges, and recent years have seen a growing integration with machine learning techniques. This review provides a comprehensive overview of how they are reshaping the field, with a particular focus on the data-driven construction of collective variables. Furthermore, these techniques have also improved biasing schemes and unlocked novel strategies via reinforcement learning and generative approaches. In addition to methodological advances, we highlight applications spanning different areas such as biomolecular processes, ligand binding, catalytic reactions, and phase transitions. We conclude by outlining future directions aimed at enabling more automated strategies for rare-event sampling.
@article{Zhu2025EnhancedApplications, author = {Zhu, Kai and Trizio, Enrico and Zhang, Jintu and Hu, Renling and Jiang, Linlong and Hou, Tingjun and Bonati, Luigi}, journal = {Chemical Reviews}, doi = {10.1021/acs.chemrev.5c00700}, month = oct, title = {Enhanced Sampling in the Age of Machine Learning: Algorithms and Applications}, year = {2025}, } - JACS
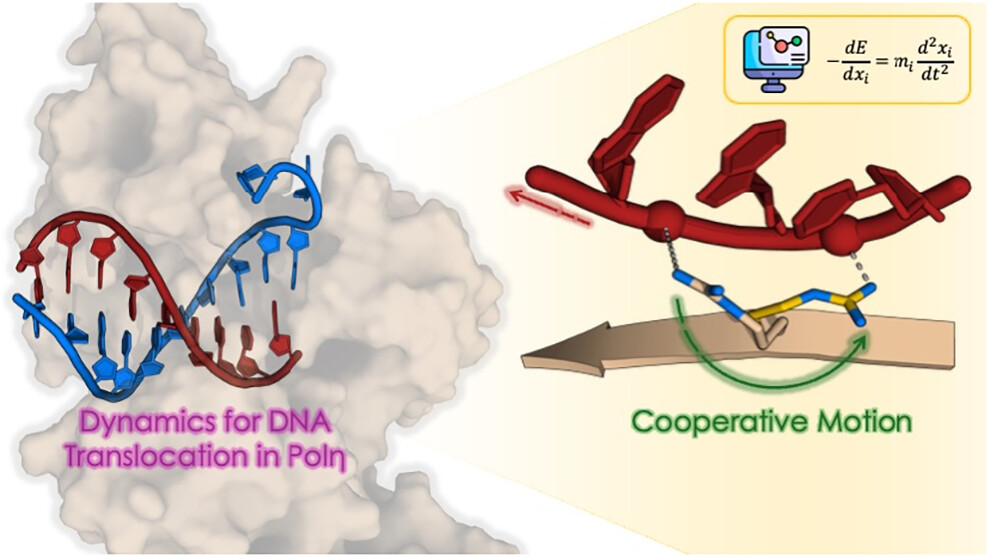 Coordinated Residue Motions at the Enzyme-Substrate Interface Promote DNA Translocation in PolymerasesAlessia Visigalli, Enrico Trizio, Luigi Bonati, Pietro Vidossich, Michele Parrinello, and Marco De VivoJournal of the American Chemical Society, Jul 2025
Coordinated Residue Motions at the Enzyme-Substrate Interface Promote DNA Translocation in PolymerasesAlessia Visigalli, Enrico Trizio, Luigi Bonati, Pietro Vidossich, Michele Parrinello, and Marco De VivoJournal of the American Chemical Society, Jul 2025The translocation of DNA in polymerase (Pol) enzymes is a critical step for Pol-mediated nucleic acid polymerization, essential for storing and transmitting genetic information in all living organisms. During this process, the newly elongated double-stranded DNA has to shift along the Pol enzyme to recreate the initial configuration at the metal-aided reactive center, where nucleotide addition can occur recurrently at every catalytic cycle. Double-stranded DNA translocation, therefore, allows the enzyme to add one more nucleotide to the growing strand, complementary to the template strand, without the enzyme dissociating from the DNA. Yet, the dynamic mechanism by which the Pol·DNA complex accomplishes DNA translocation remains poorly understood at the atomistic level. Here, leveraging recent structural data on DNA polymerase η (Polη), we elucidate its translocation mechanism, which we show to occur via an enzyme motion where the shift of Polη is asynchronous along the two DNA strands. Through equilibrium molecular dynamics and deep-learning-guided enhanced sampling simulations, we found that such a mechanism relies precisely on a set of positively charged residues of the enzyme that operate in a coordinated way at the Polη·DNA interface. Moving like screen wipers, such a dynamic mechanism of these residues promotes DNA translocation. These findings now offer new avenues to comprehend further such a complex yet fundamental dynamic process for DNA polymerization.
@article{Visigalli2025CoordinatedPolymerases, author = {Visigalli, Alessia and Trizio, Enrico and Bonati, Luigi and Vidossich, Pietro and Parrinello, Michele and Vivo, Marco De}, doi = {10.1021/jacs.5c05888}, issn = {15205126}, issue = {26}, journal = {Journal of the American Chemical Society}, month = jul, pages = {22972-22985}, pmid = {40527759}, publisher = {American Chemical Society}, title = {Coordinated Residue Motions at the Enzyme-Substrate Interface Promote DNA Translocation in Polymerases}, volume = {147}, year = {2025}, } - ACS Catal.
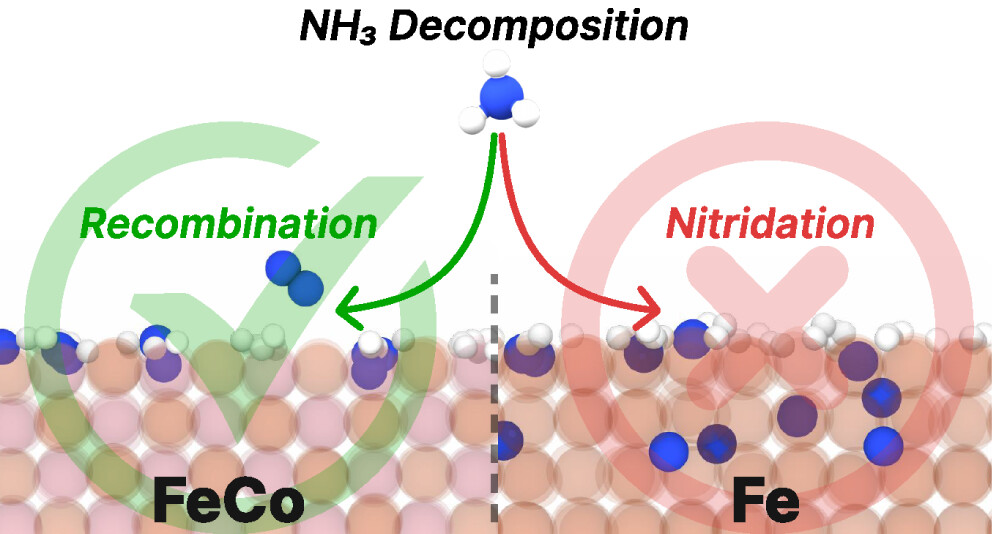 No Time for Nitrides: How Cobalt Alloying Promotes Iron Catalysts for Ammonia DecompositionSimone Perego, Maximilian Purcel, Yannick Baum, Shilong Chen, Astrid Sophie Müller, Michele Parrinello, Malte Behrens, Martin Muhler, and Luigi BonatiACS Catalysis, Oct 2025
No Time for Nitrides: How Cobalt Alloying Promotes Iron Catalysts for Ammonia DecompositionSimone Perego, Maximilian Purcel, Yannick Baum, Shilong Chen, Astrid Sophie Müller, Michele Parrinello, Malte Behrens, Martin Muhler, and Luigi BonatiACS Catalysis, Oct 2025The increasing demand for hydrogen production has driven interest in ammonia decomposition. Iron-based catalysts, widely used for ammonia synthesis, exhibit suboptimal performance in the reverse pr...
@article{Perego2025NoDecomposition, author = {Perego, Simone and Purcel, Maximilian and Baum, Yannick and Chen, Shilong and Müller, Astrid Sophie and Parrinello, Michele and Behrens, Malte and Muhler, Martin and Bonati, Luigi}, doi = {10.1021/ACSCATAL.5C04795}, issn = {2155-5435}, issue = {19}, journal = {ACS Catalysis}, keywords = {alloying,ammonia decomposition,machine learning potentials,molecular dynamics,nitridation}, month = oct, pages = {16690-16702}, publisher = {American Chemical Society}, title = {No Time for Nitrides: How Cobalt Alloying Promotes Iron Catalysts for Ammonia Decomposition}, volume = {15}, url = {/doi/pdf/10.1021/acscatal.5c04795?ref=article_openPDF}, year = {2025}, } - J. Cat.
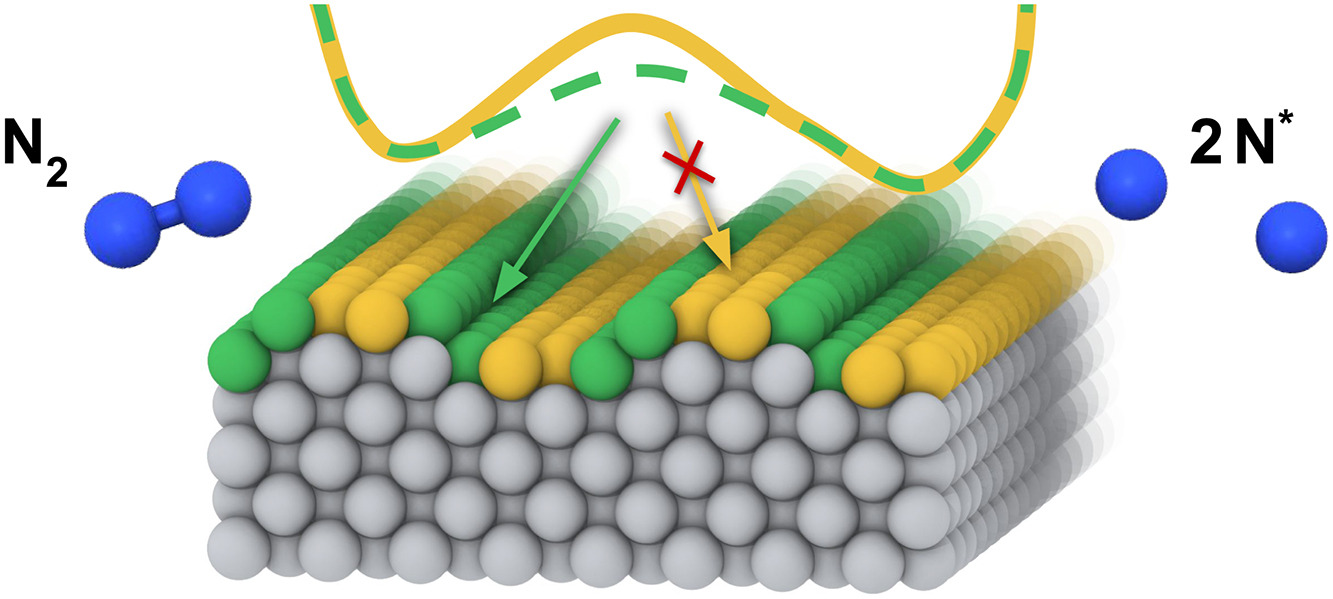 Nitrogen adsorption and dissociation on flat and stepped Fe (110) surfacesShivam Tripathi, Luigi Bonati, Simone Perego, and Michele ParrinelloJournal of Catalysis, Nov 2025
Nitrogen adsorption and dissociation on flat and stepped Fe (110) surfacesShivam Tripathi, Luigi Bonati, Simone Perego, and Michele ParrinelloJournal of Catalysis, Nov 2025Stepped surfaces are known to exhibit higher catalytic activity than flat ones in ammonia synthesis, yet an atomistic-level understanding remains elusive. In this work, we employ molecular dynamics simulations powered by machine learning potentials combined with enhanced sampling techniques to study the initial stages of the Haber–Bosch process, namely the dissociative chemisorption of N2 under operando temperature conditions. We compare a flat Fe(110) surface with a series of stepped Fe(110) surfaces of increasing step density. Our results show that the presence of steps significantly lowers the dissociation barrier. Importantly, the catalytic enhancement is not correlated with the step density but stems from the intrinsic reactivity of the step sites themselves. We further investigate the fate of the dissociated nitrogen atoms by computing escape times from the step edges, allowing us to assess the likelihood and timescale of step poisoning.
- 2D Mat.
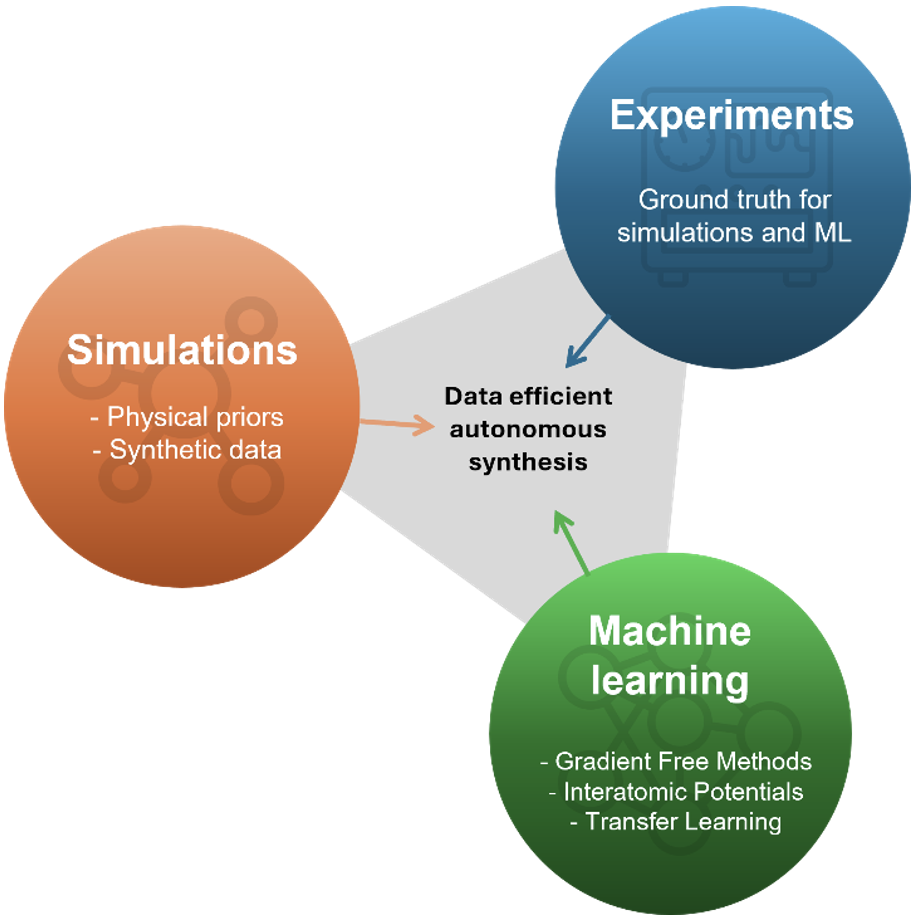 From trial-and-error to intelligent workflows: machine learning and simulations for scalable 2D material synthesisLuigi Bonati, Pietro Novelli, and Antonio Rossi2D Materials, Dec 2025
From trial-and-error to intelligent workflows: machine learning and simulations for scalable 2D material synthesisLuigi Bonati, Pietro Novelli, and Antonio Rossi2D Materials, Dec 2025Wafer-scale growth of two-dimensional (2D) materials remains an unsolved challenge. Current synthesis still depends on trial-and-error, limiting reproducibility and slowing progress toward practical technologies. This perspective argues that machine learning (ML) and atomistic simulations can provide the foundation for intelligent synthesis workflows. Gradient-free optimization methods, including Bayesian optimization and adaptive Monte Carlo, already demonstrated that high-quality growth conditions can be learned efficiently from experimental feedback. At the same time, atomistic simulations deliver physical priors, synthetic data, and mechanistic insights that focus the search and mitigate data scarcity. We propose a synergistic framework where experiments, simulations, and ML form a closed loop: experiments provide validation, simulations constrain exploration, and ML uncovers patterns and proposes new recipes. Embedding this intelligence into growth protocols can turn 2D materials synthesis into a reproducible and scalable methodology, moving beyond intuition-driven experimentation, accelerating their translation into wafer-scale technologies. While current demonstrations remain system-specific, this perspective provides a pathway toward wafer-scale technologies and data-driven discovery.
- arXivElectrochemical Interfaces at Constant Potential: Data-Efficient Transfer Learning for Machine-Learning-Based Molecular DynamicsMichele Giovanni Bianchi, Michele Re Fiorentin, Francesca Risplendi, Candido Fabrizio Pirri, Michele Parrinello, Luigi Bonati, and Giancarlo CiceroarXiv preprint arXiv:2511.19338, Nov 2025
Simulating electrified metal/water interfaces with explicit solvent under constant potential is essential for understanding electrochemical processes, yet remains prohibitively expensive with ab initio methods. We present TRECI, a data-efficient workflow for constructing machine learning force-fields (ML-FFs) that achieve ab initio-level accuracy in electronically grand-canonical molecular dynamics. By leveraging transfer learning from general-purpose and domain-specific models, TRECI enables stable and accurate simulations across a wide potential range using a reduced number of reference configurations. This efficiency allows the use of high-level meta-GGA functionals and rigorous surface-electrification schemes. Applied to Cu(111)/water, models trained on just one thousand configurations yield accurate molecular dynamics simulations, capturing bias-dependent solvent restructuring effects not previously reported. TRECI offers a general strategy for characterising diverse materials and interfacial chemistries, significantly lowering the cost of realistic constant-potential simulations and expanding access to quantitative electrochemical modelling.
2024
- npj Comput. Mater.
 Data efficient machine learning potentials for modeling catalytic reactivity via active learning and enhanced samplingSimone Perego and Luigi Bonatinpj Computational Materials, Dec 2024
Data efficient machine learning potentials for modeling catalytic reactivity via active learning and enhanced samplingSimone Perego and Luigi Bonatinpj Computational Materials, Dec 2024Simulating catalytic reactivity under operative conditions poses a significant challenge due to the dynamic nature of the catalysts and the high computational cost of electronic structure calculations. Machine learning potentials offer a promising avenue to simulate dynamics at a fraction of the cost, but they require datasets containing all relevant configurations, particularly reactive ones. Here, we present a scheme to construct reactive potentials in a data-efficient manner. This is achieved by combining enhanced sampling methods first with Gaussian processes to discover transition paths and then with graph neural networks to obtain a uniformly accurate description. The necessary configurations are extracted via a Data-Efficient Active Learning (DEAL) procedure based on local environment uncertainty. We validated our approach by studying several reactions related to the decomposition of ammonia on iron-cobalt alloy catalysts. Our scheme proved to be efficient, requiring only 1000 DFT calculations per reaction, and robust, sampling reactive configurations from the different accessible pathways. Using this potential, we calculated free energy profiles and characterized reaction mechanisms, showing the ability to provide microscopic insights into complex processes under dynamic conditions.
@article{Perego2024DataSampling, author = {Perego, Simone and Bonati, Luigi}, doi = {10.1038/S41524-024-01481-6}, issn = {20573960}, issue = {1}, journal = {npj Computational Materials}, keywords = {Atomistic models,Computational methods,Heterogeneous catalysis,Theoretical chemistry}, month = dec, pages = {1-13}, publisher = {Nature Research}, title = {Data efficient machine learning potentials for modeling catalytic reactivity via active learning and enhanced sampling}, volume = {10}, url = {https://www.nature.com/articles/s41524-024-01481-6}, year = {2024}, } - JCTC
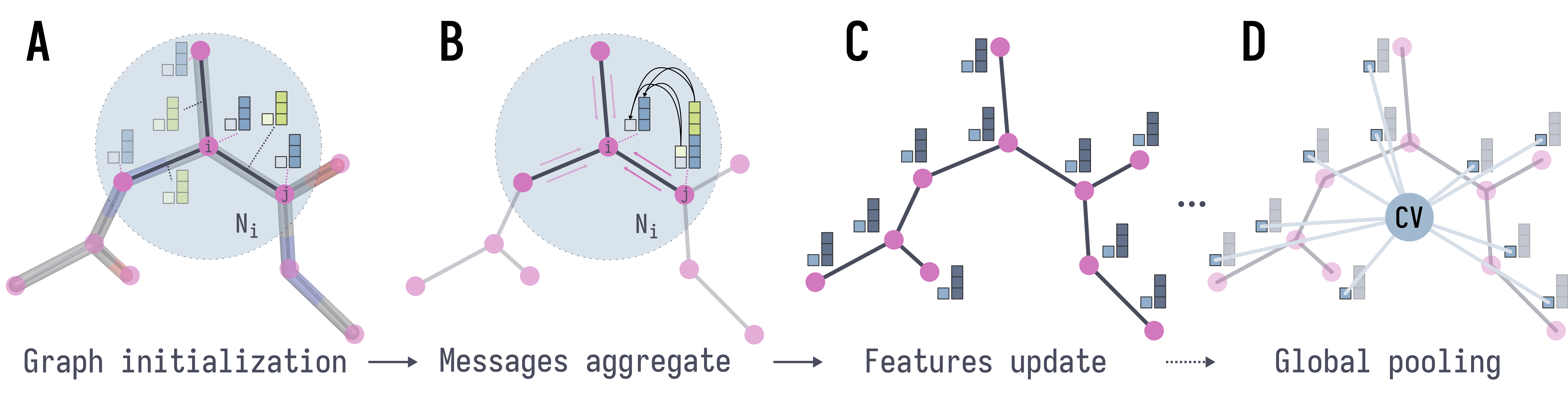 Descriptor-Free Collective Variables from Geometric Graph Neural NetworksJintu Zhang, Luigi Bonati, Enrico Trizio, Odin Zhang, Yu Kang, Ting Jun Hou, and Michele ParrinelloJournal of Chemical Theory and Computation, Dec 2024
Descriptor-Free Collective Variables from Geometric Graph Neural NetworksJintu Zhang, Luigi Bonati, Enrico Trizio, Odin Zhang, Yu Kang, Ting Jun Hou, and Michele ParrinelloJournal of Chemical Theory and Computation, Dec 2024Enhanced sampling simulations make the computational study of rare events feasible. A large family of such methods crucially depends on the definition of some collective variables (CVs) that could ...
@article{Zhang2024DescriptorFreeNetworks, author = {Zhang, Jintu and Bonati, Luigi and Trizio, Enrico and Zhang, Odin and Kang, Yu and Hou, Ting Jun and Parrinello, Michele}, doi = {10.1021/ACS.JCTC.4C01197}, issn = {15499626}, issue = {24}, journal = {Journal of Chemical Theory and Computation}, month = dec, pages = {10787-10797}, pmid = {39665183}, publisher = {American Chemical Society}, title = {Descriptor-Free Collective Variables from Geometric Graph Neural Networks}, volume = {20}, url = {/doi/pdf/10.1021/acs.jctc.4c01197}, year = {2024}, } - arXivAdvanced simulations with PLUMED: OPES and Machine Learning Collective VariablesEnrico Trizio, Andrea Rizzi, Pablo M. Piaggi, Michele Invernizzi, and Luigi BonatiarXiv, Oct 2024
Many biological processes occur on time scales longer than those accessible to molecular dynamics simulations. Identifying collective variables (CVs) and introducing an external potential to accelerate them is a popular approach to address this problem. In particular, \texttt{PLUMED} is a community-developed library that implements several methods for CV-based enhanced sampling. This chapter discusses two recent developments that have gained popularity in recent years. The first is the On-the-fly Probability Enhanced Sampling (OPES) method as a biasing scheme. This provides a unified approach to enhanced sampling able to cover many different scenarios: from free energy convergence to the discovery of metastable states, from rate calculation to generalized ensemble simulation. The second development concerns the use of machine learning (ML) approaches to determine CVs by learning the relevant variables directly from simulation data. The construction of these variables is facilitated by the \texttt{mlcolvar} library, which allows them to be optimized in Python and then used to enhance sampling thanks to a native interface inside \texttt{PLUMED}. For each of these methods, in addition to a brief introduction, we provide guidelines, practical suggestions and point to examples from the literature to facilitate their use in the study of the process of interest.
@article{Trizio2024AdvancedVariables, author = {Trizio, Enrico and Rizzi, Andrea and Piaggi, Pablo M. and Invernizzi, Michele and Bonati, Luigi}, isbn = {2410.18019v1}, journal = {arXiv}, keywords = {Enhanced sampling,OPES,PLUMED,collective variables,machine learning,mlcolvar}, month = oct, title = {Advanced simulations with PLUMED: OPES and Machine Learning Collective Variables}, url = {http://arxiv.org/abs/2410.18019}, year = {2024}, } - ACS Catal.
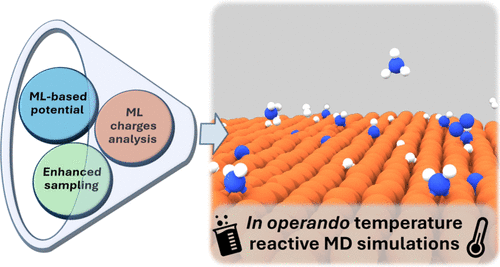 How Dynamics Changes Ammonia Cracking on Iron SurfacesSimone Perego, Luigi Bonati, Shivam Tripathi, and Michele ParrinelloACS Catalysis, Oct 2024
How Dynamics Changes Ammonia Cracking on Iron SurfacesSimone Perego, Luigi Bonati, Shivam Tripathi, and Michele ParrinelloACS Catalysis, Oct 2024Being rich in hydrogen and easy to transport, ammonia is a promising hydrogen carrier. However, a microscopic characterization of the ammonia cracking reaction is still lacking, hindered by extreme operando conditions. Leveraging state-of-the-art molecular dynamics, machine learning potentials, and enhanced sampling methods, we offer an atomistic view of the adsorption, diffusion, and dehydrogenation processes of a single NHx (x = 1, 3) molecule on two representative surfaces at the operando temperature of 700 K. We elucidate the effects of the dynamics on all the steps of decomposition. On the stable (110) surface, we found that the reaction intermediate diffusions are favored over dehydrogenation, with non-negligible effects on the reactivity for one intermediate. The role is even more dramatic on the (111) surface, where the mobility of Fe surface atoms introduces unexplored adsorption sites and significantly alters the dehydrogenation barriers. In both cases, a detailed analysis of reactive events shows that there is never a single transition state, but it is always an ensemble. Notwithstanding, a unified mechanism can be identified by following the charge transfer along the different reaction pathways.
@article{Perego2024HowSurfaces, author = {Perego, Simone and Bonati, Luigi and Tripathi, Shivam and Parrinello, Michele}, doi = {10.1021/ACSCATAL.4C01920}, issn = {21555435}, issue = {19}, journal = {ACS Catalysis}, keywords = {ammonia decomposition,dynamics,enhanced sampling,green hydrogen,heterogeneous catalysis,machine learning,molecular dynamics,neural network potential}, month = oct, pages = {14652-14664}, publisher = {American Chemical Society}, title = {How Dynamics Changes Ammonia Cracking on Iron Surfaces}, volume = {14}, url = {/doi/pdf/10.1021/acscatal.4c01920?ref=article_openPDF}, year = {2024}, } - ACS Catal.
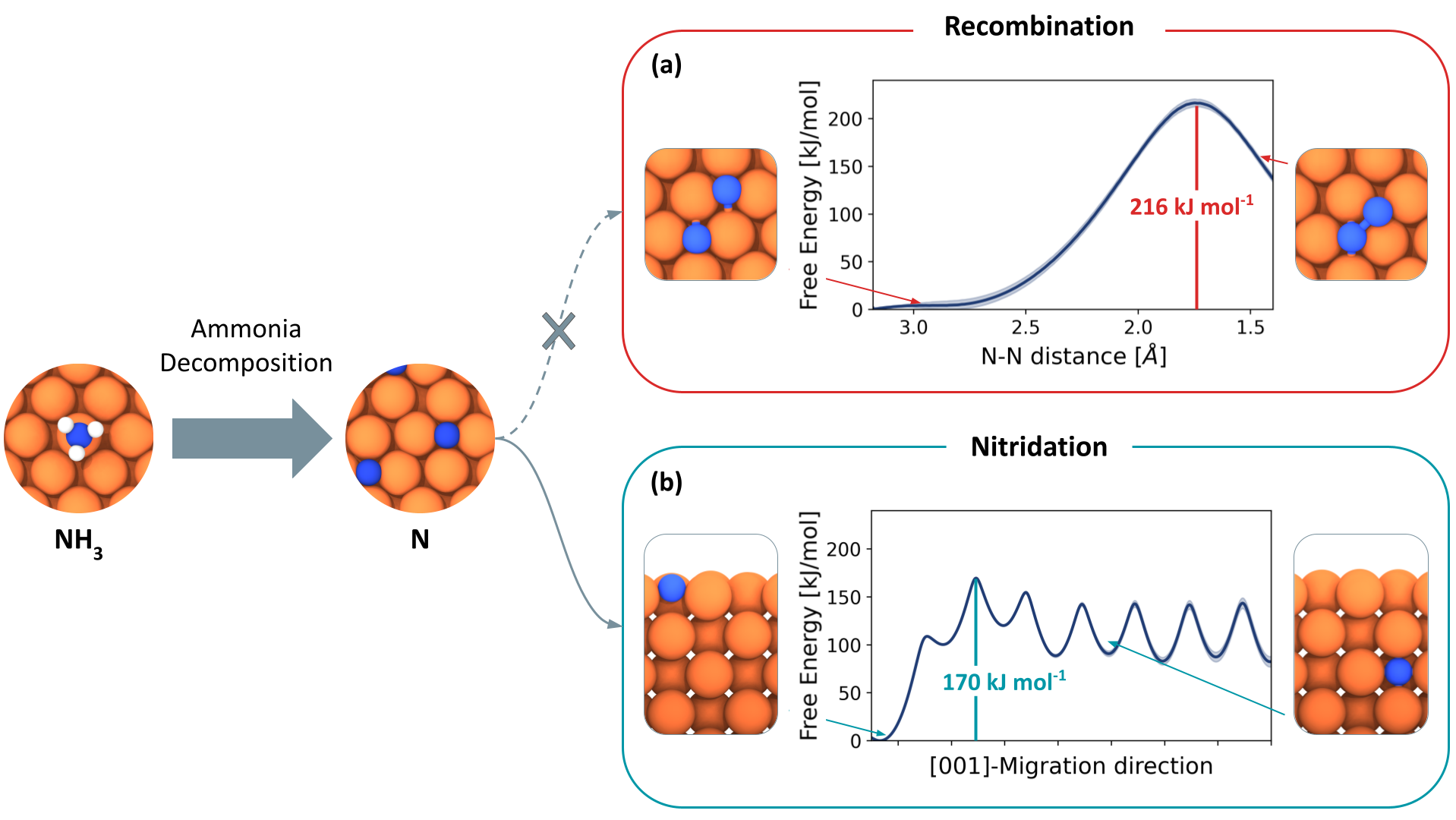 Iron Nitride Formation and Decomposition during Ammonia Decomposition over a Wustite-Based Bulk Iron CatalystM. Purcel, S. Berendts, L. Bonati, S. Perego, A. Müller, M. Lerch, M. Parrinello, and M. MuhlerACS Catalysis, Sep 2024
Iron Nitride Formation and Decomposition during Ammonia Decomposition over a Wustite-Based Bulk Iron CatalystM. Purcel, S. Berendts, L. Bonati, S. Perego, A. Müller, M. Lerch, M. Parrinello, and M. MuhlerACS Catalysis, Sep 2024Hydrogen production using renewable sources shows great promise in lowering the dependence on fossil fuels in our current energy system, but challenges persist in its storage and transportation. Thus, ammonia emerges as a viable carrier for H2 due to its favorable properties. NH3 decomposition was studied at atmospheric pressure over a multiply promoted fused FeO-based bulk catalyst optimized for NH3 synthesis by applying transient kinetic experiments, in situ XRD, and Molecular dynamics (MD) simulations. Transient NH3 decomposition and temperature-programmed experiments in a fixed-bed reactor yielded the activation energies of bulk iron nitride formation and decomposition, which occurred in two steps: Fe ⇌ Fe4N ⇌ Fe2N. The decomposition of Fe4N into Fe and N2 was identified as the rate-determining step of NH3 decomposition with essentially the same activation energy amounting to 172 and 173 kJ mol-1, respectively. Fe4N and Fe2N were identified based on the nitrogen mass balance and confirmed as γ′-Fe4N and ϵ-Fe3N1.5 by in situ XRD experiments in NH3. MD simulations showed that the migration of adsorbed N atoms into the bulk of the catalyst is favored over recombinative desorption, influencing N storage and N2 release dynamics. Overall, the findings contribute to a comprehensive understanding of NH3 synthesis catalysts under decomposition conditions, identifying the key descriptor to enhance the performance of Fe-based catalysts.
@article{Purcel2024IronCatalyst, author = {Purcel, M. and Berendts, S. and Bonati, L. and Perego, S. and Müller, A. and Lerch, M. and Parrinello, M. and Muhler, M.}, doi = {10.1021/ACSCATAL.4C04415}, issn = {21555435}, issue = {18}, journal = {ACS Catalysis}, keywords = {ammonia decomposition,bulk iron catalyst,in situ XRD,machine learning,molecular dynamics simulations,nitridation,transient kinetics}, month = sep, pages = {13947-13957}, publisher = {American Chemical Society}, title = {Iron Nitride Formation and Decomposition during Ammonia Decomposition over a Wustite-Based Bulk Iron Catalyst}, volume = {14}, url = {/doi/pdf/10.1021/acscatal.4c04415?ref=article_openPDF}, year = {2024}, } - JCTC
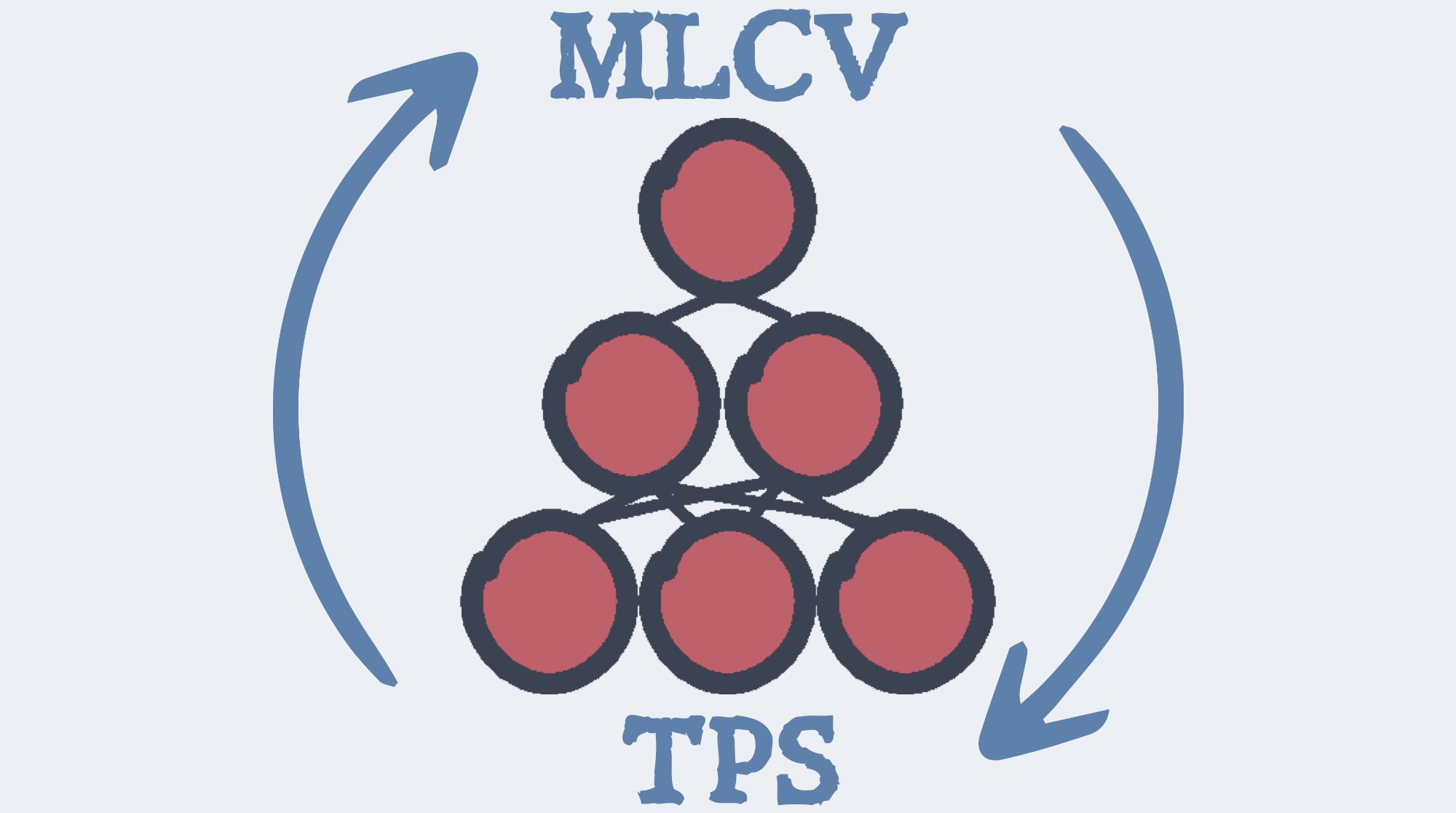 Combining Transition Path Sampling with Data-Driven Collective Variables through a Reactivity-Biased Shooting AlgorithmJintu Zhang, Odin Zhang, Luigi Bonati, and Ting Jun HouJournal of Chemical Theory and Computation, Jun 2024
Combining Transition Path Sampling with Data-Driven Collective Variables through a Reactivity-Biased Shooting AlgorithmJintu Zhang, Odin Zhang, Luigi Bonati, and Ting Jun HouJournal of Chemical Theory and Computation, Jun 2024Rare event sampling is a central problem in modern computational chemistry research. Among the existing methods, transition path sampling (TPS) can generate unbiased representations of reaction pro...
@article{Zhang2024CombiningAlgorithm, author = {Zhang, Jintu and Zhang, Odin and Bonati, Luigi and Hou, Ting Jun}, doi = {10.1021/ACS.JCTC.4C00423}, issn = {15499626}, issue = {11}, journal = {Journal of Chemical Theory and Computation}, month = jun, pages = {4523-4532}, pmid = {38801759}, publisher = {American Chemical Society}, title = {Combining Transition Path Sampling with Data-Driven Collective Variables through a Reactivity-Biased Shooting Algorithm}, volume = {20}, year = {2024}, } - JCP
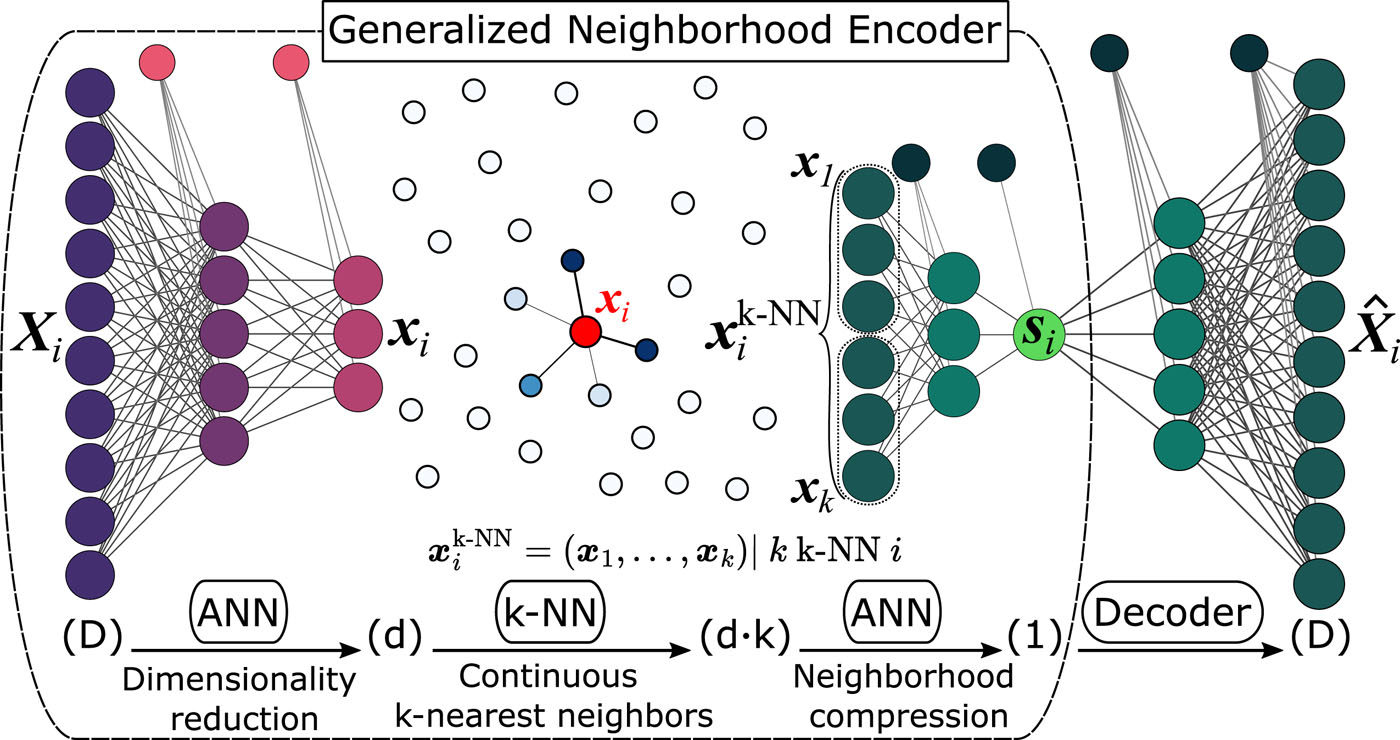 Deep learning path-like collective variable for enhanced sampling molecular dynamicsThorben Fröhlking, Luigi Bonati, Valerio Rizzi, and Francesco Luigi GervasioThe Journal of Chemical Physics, May 2024
Deep learning path-like collective variable for enhanced sampling molecular dynamicsThorben Fröhlking, Luigi Bonati, Valerio Rizzi, and Francesco Luigi GervasioThe Journal of Chemical Physics, May 2024Several enhanced sampling techniques rely on the definition of collective variables to effectively explore free energy landscapes. The existing variables that describe the progression along a reactive pathway offer an elegant solution but face a number of limitations. In this paper, we address these challenges by introducing a new path-like collective variable called the “deep-locally non-linear-embedding,” which is inspired by principles of the locally linear embedding technique and is trained on a reactive trajectory. The variable mimics the ideal reaction coordinate by automatically generating a non-linear combination of features through a differentiable generalized autoencoder that combines a neural network with a continuous k-nearest neighbor selection. Among the key advantages of this method is its capability to automatically choose the metric for searching neighbors and to learn the path from state A to state B without the need to handpick landmarks a priori. We demonstrate the effectiveness of DeepLNE by showing that the progression along the path variable closely approximates the ideal reaction coordinate in toy models, such as the Müller-Brown potential and alanine dipeptide. Then, we use it in the molecular dynamics simulations of an RNA tetraloop, where we highlight its capability to accelerate transitions and estimate the free energy of folding.
@article{Frohlking2024DeepDynamics, author = {Fröhlking, Thorben and Bonati, Luigi and Rizzi, Valerio and Gervasio, Francesco Luigi}, doi = {10.1063/5.0202156}, issn = {0021-9606}, issue = {17}, journal = {The Journal of Chemical Physics}, month = may, pages = {174109}, pmid = {38748013}, publisher = {AIP Publishing}, title = {Deep learning path-like collective variable for enhanced sampling molecular dynamics}, volume = {160}, url = {/aip/jcp/article/160/17/174109/3287814/Deep-learning-path-like-collective-variable-for}, year = {2024}, } - ACS Catal.
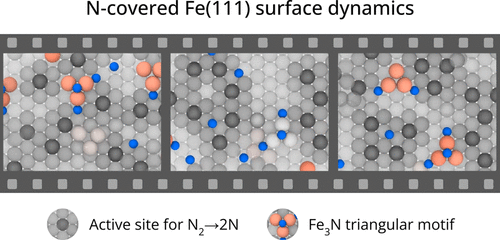 How Poisoning Is Avoided in a Step of Relevance to the Haber-Bosch CatalysisShivam Tripathi, Luigi Bonati, Simone Perego, and Michele ParrinelloACS Catalysis , Apr 2024
How Poisoning Is Avoided in a Step of Relevance to the Haber-Bosch CatalysisShivam Tripathi, Luigi Bonati, Simone Perego, and Michele ParrinelloACS Catalysis , Apr 2024For a catalyst to be efficient and durable, it is crucial that the reaction products do not poison the catalyst. In the case of the Haber-Bosch process, the rate-limiting step is believed to be the decomposition of nitrogen molecules on the Fe(111) surface. This step leads to the production on the surface of atomic nitrogen (N*), which, unless hydrogenated and eventually released as ammonia, remains adsorbed and occupies the active sites. Thus, it is important to ascertain how a high N* coverage affects the nitrogen dissociative chemisorption. To answer this question, we study the properties of the Fe(111) surface at different N* coverage both at room and operando temperature. In the latter regime, we have already found that Fe surface atoms exhibit a high mobility, promoting the formation of adatoms and vacancies, and causing the catalytic centers to acquire a finite lifetime [Bonati et al. Proceedings of the National Academy of Sciences 2023, 120 (50), e2313023120 ]. We discover that the N* coverage reduces but does not eliminate the iron mobility. Remarkably, the N* atoms stabilize triangular surface structures associated with the formation of vacancies, which are a sign of a frustrated drive toward a more stable Fe4N phase. As a consequence, nitrogen atoms tend to cluster, reducing their poisoning effect. At the same time, the reduction in the number of catalytic centers is counteracted by an increase in their lifetime. The combined effect is that the dissociation barrier is not significantly altered in the range of coverages studied. These results bring to light the complex role that dynamics plays in catalytic reactivity under operando conditions.
@article{Tripathi2024HowCatalysis, author = {Tripathi, Shivam and Bonati, Luigi and Perego, Simone and Parrinello, Michele}, doi = {10.1021/ACSCATAL.3C06201}, issn = {21555435}, issue = {7}, journal = {ACS Catalysis }, keywords = {Haber−Bosch,dissociative chemisorption,iron (111),neural network potential,operando conditions,poisoning,surface dynamics}, month = apr, pages = {4944-4950}, publisher = {American Chemical Society}, title = {How Poisoning Is Avoided in a Step of Relevance to the Haber-Bosch Catalysis}, volume = {14}, url = {/doi/pdf/10.1021/acscatal.3c06201?ref=article_openPDF}, year = {2024}, } - npj Comput. Mater.
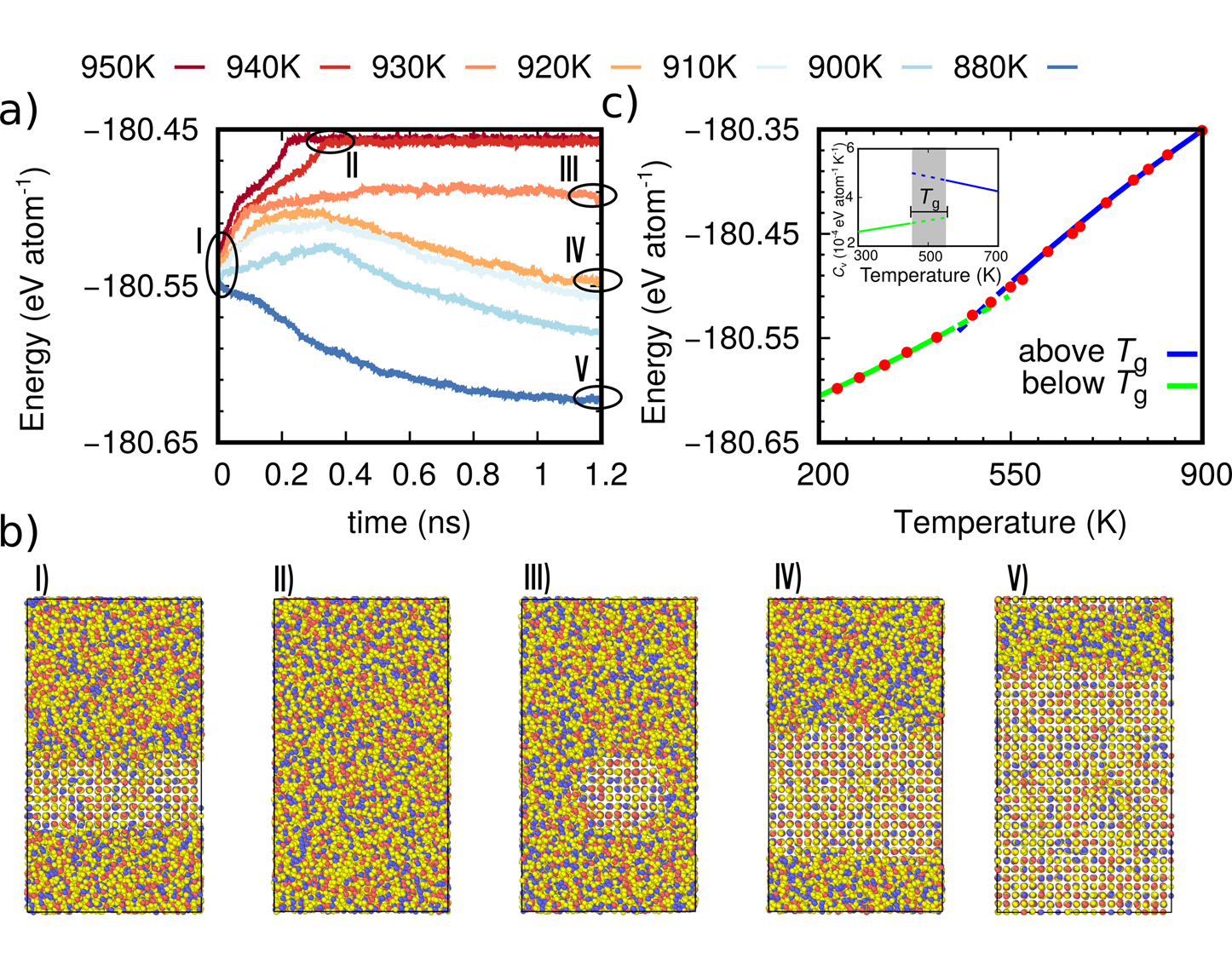 Unraveling the crystallization kinetics of the Ge2Sb2Te5 phase change compound with a machine-learned interatomic potentialOmar Abou El Kheir, Luigi Bonati, Michele Parrinello, and Marco Bernasconinpj Computational Materials, Dec 2024
Unraveling the crystallization kinetics of the Ge2Sb2Te5 phase change compound with a machine-learned interatomic potentialOmar Abou El Kheir, Luigi Bonati, Michele Parrinello, and Marco Bernasconinpj Computational Materials, Dec 2024The phase change compound Ge2Sb2Te5 (GST225) is exploited in advanced non-volatile electronic memories and in neuromorphic devices which both rely on a fast and reversible transition between the crystalline and amorphous phases induced by Joule heating. The crystallization kinetics of GST225 is a key functional feature for the operation of these devices. We report here on the development of a machine-learned interatomic potential for GST225 that allowed us to perform large scale molecular dynamics simulations (over 10,000 atoms for over 100 ns) to uncover the details of the crystallization kinetics in a wide range of temperatures of interest for the programming of the devices. The potential is obtained by fitting with a deep neural network (NN) scheme a large quantum-mechanical database generated within density functional theory. The availability of a highly efficient and yet highly accurate NN potential opens the possibility to simulate phase change materials at the length and time scales of the real devices.
@article{AbouElKheir2024UnravelingPotential, author = {Kheir, Omar Abou El and Bonati, Luigi and Parrinello, Michele and Bernasconi, Marco}, doi = {10.1038/S41524-024-01217-6}, issn = {20573960}, issue = {1}, journal = {npj Computational Materials}, keywords = {Atomistic models,Electronic devices,Structure of solids and liquids}, month = dec, pages = {1-12}, publisher = {Nature Research}, title = {Unraveling the crystallization kinetics of the Ge2Sb2Te5 phase change compound with a machine-learned interatomic potential}, volume = {10}, url = {https://www.nature.com/articles/s41524-024-01217-6}, year = {2024}, }
2023
- JCP
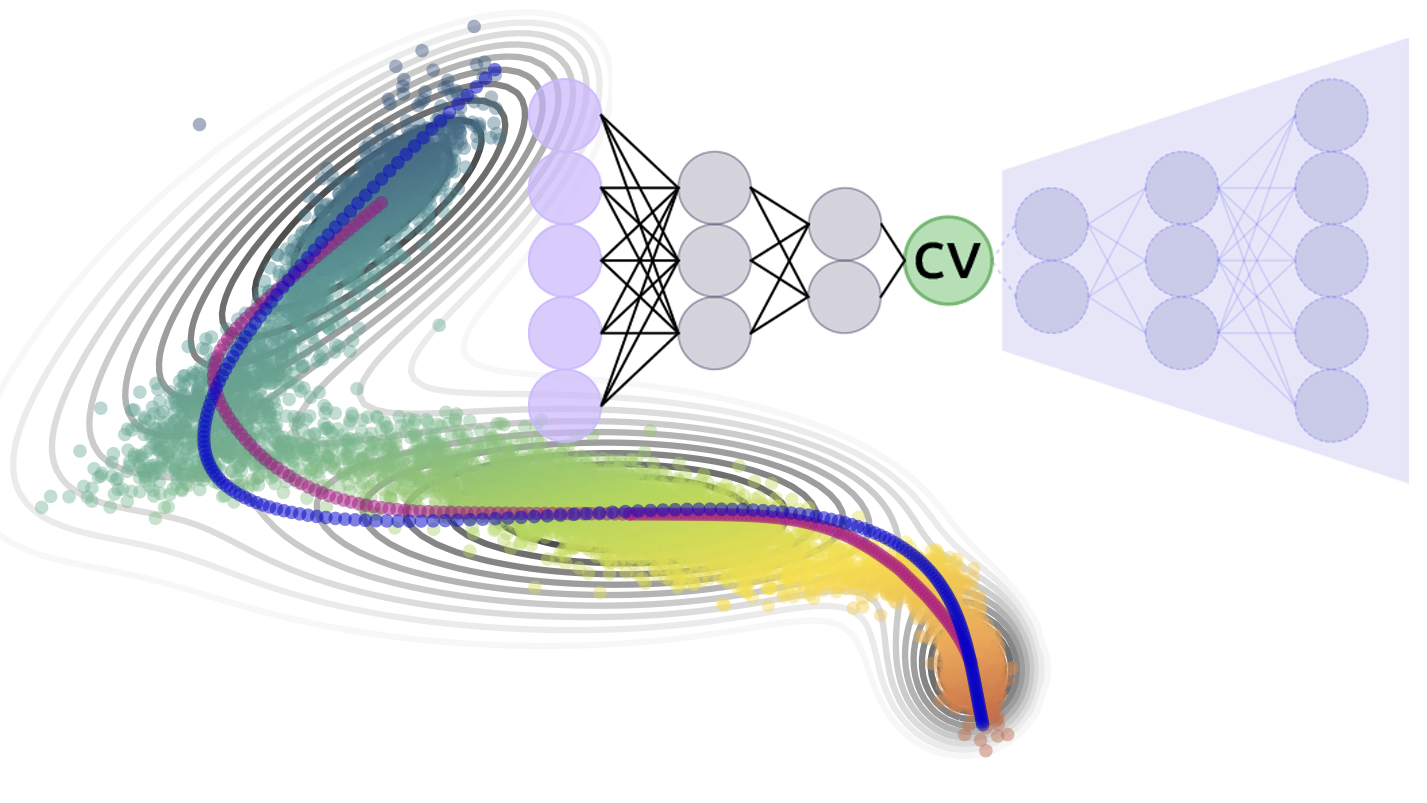 A unified framework for machine learning collective variables for enhanced sampling simulations: mlcolvarLuigi Bonati, Enrico Trizio, Andrea Rizzi, and Michele ParrinelloThe Journal of Chemical Physics, Jul 2023
A unified framework for machine learning collective variables for enhanced sampling simulations: mlcolvarLuigi Bonati, Enrico Trizio, Andrea Rizzi, and Michele ParrinelloThe Journal of Chemical Physics, Jul 2023Identifying a reduced set of collective variables is critical for understanding atomistic simulations and accelerating them through enhanced sampling techniques. Recently, several methods have been proposed to learn these variables directly from atomistic data. Depending on the type of data available, the learning process can be framed as dimensionality reduction, classification of metastable states, or identification of slow modes. Here, we present mlcolvar, a Python library that simplifies the construction of these variables and their use in the context of enhanced sampling through a contributed interface to the PLUMED software. The library is organized modularly to facilitate the extension and cross-contamination of these methodologies. In this spirit, we developed a general multi-task learning framework in which multiple objective functions and data from different simulations can be combined to improve the collective variables. The library’s versatility is demonstrated through simple examples that are prototypical of realistic scenarios.
@article{Bonati2023Amlcolvar, author = {Bonati, Luigi and Trizio, Enrico and Rizzi, Andrea and Parrinello, Michele}, doi = {10.1063/5.0156343}, issn = {0021-9606}, issue = {1}, journal = {The Journal of Chemical Physics}, month = jul, pmid = {37409767}, publisher = {AIP Publishing}, title = {A unified framework for machine learning collective variables for enhanced sampling simulations: <tt>mlcolvar</tt>}, volume = {159}, url = {https://pubs.aip.org/jcp/article/159/1/014801/2901354/A-unified-framework-for-machine-learning}, year = {2023}, } - Neurips
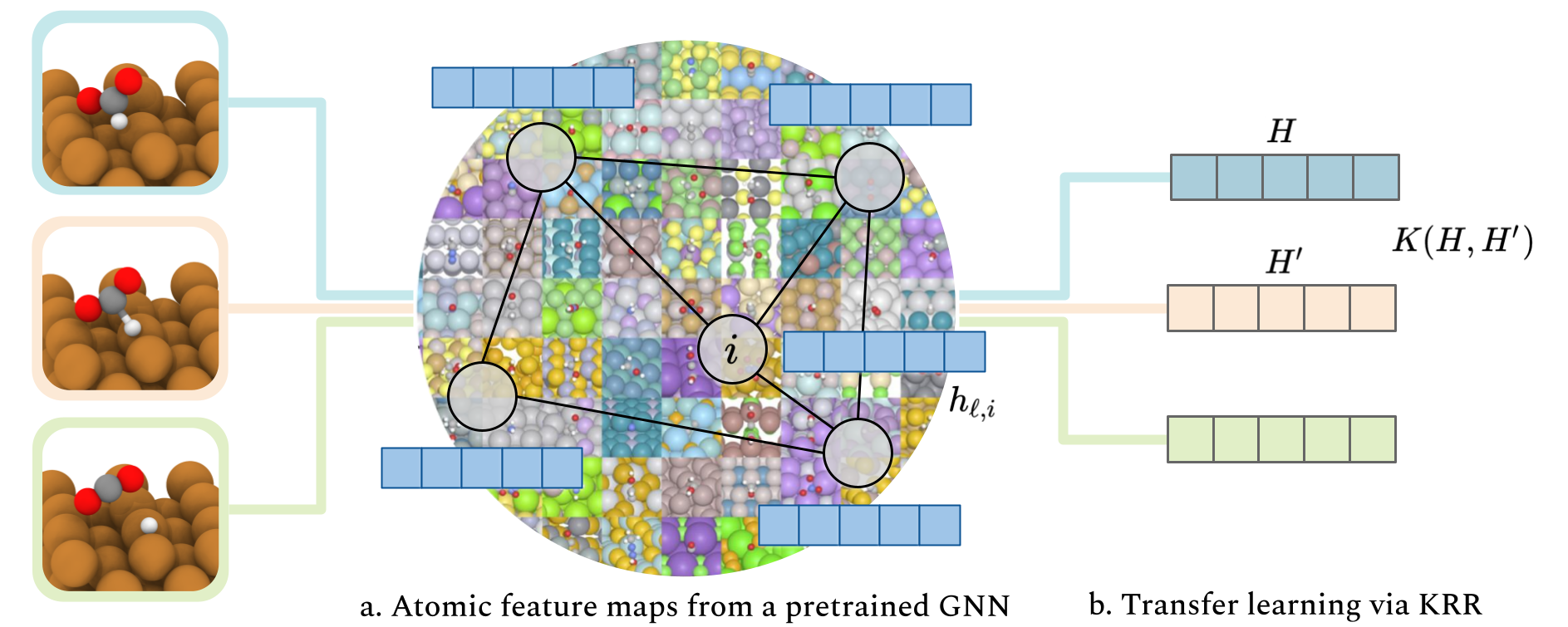 Transfer learning for atomistic simulations using GNNs and kernel mean embeddingsJohn Falk, Luigi Bonati, Pietro Novelli, Michele Parrinello, and Massimiliano PontilAdvances in Neural Information Processing Systems, Jul 2023
Transfer learning for atomistic simulations using GNNs and kernel mean embeddingsJohn Falk, Luigi Bonati, Pietro Novelli, Michele Parrinello, and Massimiliano PontilAdvances in Neural Information Processing Systems, Jul 2023@article{Falk2023Transfer, title = {Transfer learning for atomistic simulations using GNNs and kernel mean embeddings}, author = {Falk, John and Bonati, Luigi and Novelli, Pietro and Parrinello, Michele and Pontil, Massimiliano}, journal = {Advances in Neural Information Processing Systems}, volume = {36}, pages = {29783--29797}, year = {2023}, } - PNAS
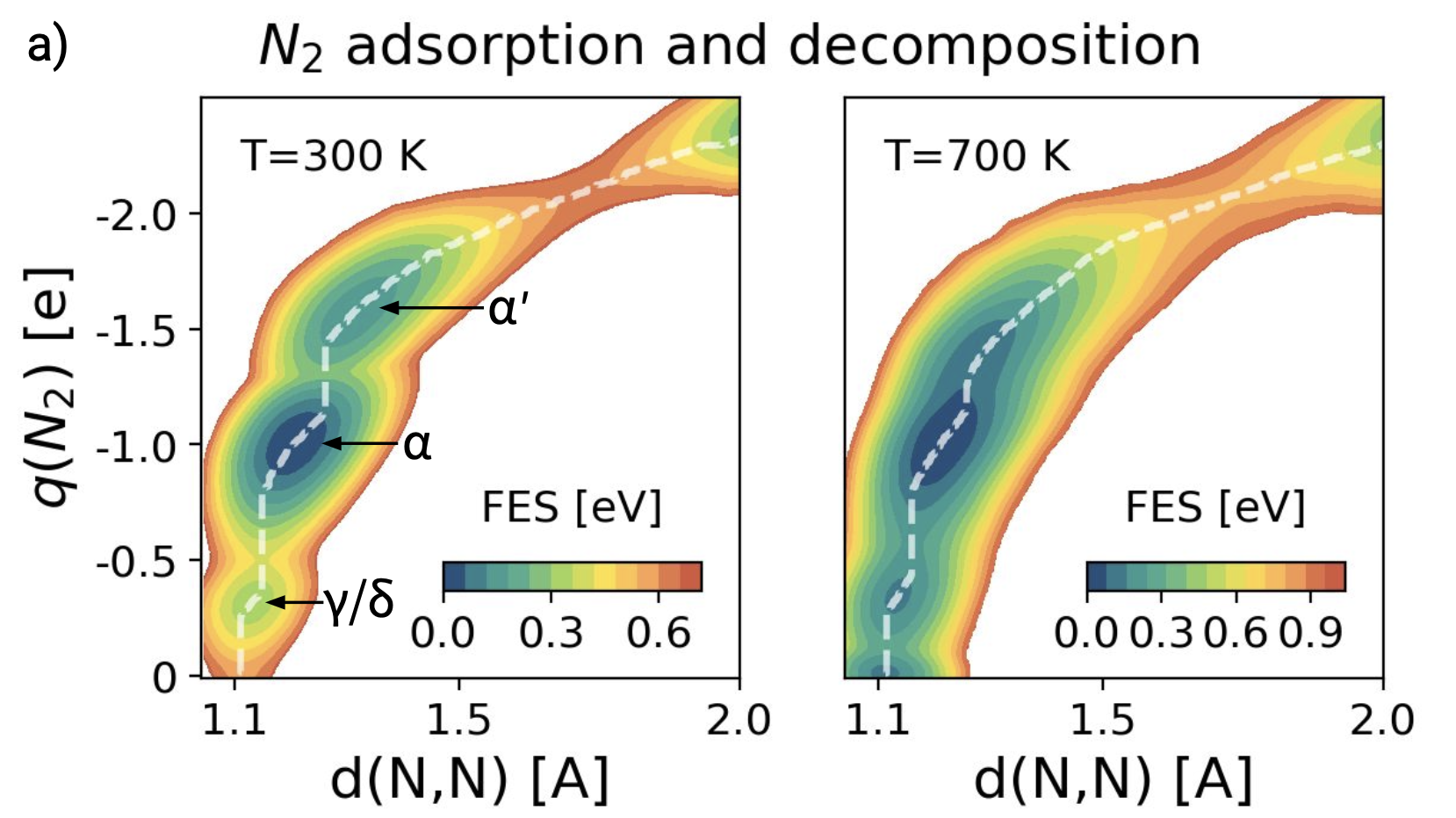 The role of dynamics in heterogeneous catalysis: Surface diffusivity and N2 decomposition on Fe(111)Luigi Bonati, Daniela Polino, Cristina Pizzolitto, Pierdomenico Biasi, Rene Eckert, Stephan Reitmeier, Robert Schlögl, and Michele ParrinelloProceedings of the National Academy of Sciences of the United States of America, Dec 2023
The role of dynamics in heterogeneous catalysis: Surface diffusivity and N2 decomposition on Fe(111)Luigi Bonati, Daniela Polino, Cristina Pizzolitto, Pierdomenico Biasi, Rene Eckert, Stephan Reitmeier, Robert Schlögl, and Michele ParrinelloProceedings of the National Academy of Sciences of the United States of America, Dec 2023Dynamics has long been recognized to play an important role in heterogeneous catalytic processes. However, until recently, it has been impossible to study their dynamical behavior at industry-relevant temperatures. Using a combination of machine learning potentials and advanced simulation techniques, we investigate the cleavage of the N2 triple bond on the Fe(111) surface. We find that at low temperatures our results agree with the well-established picture. However, if we increase the temperature to reach operando conditions, the surface undergoes a global dynamical change and the step structure of the Fe(111) surface is destabilized. The catalytic sites, traditionally associated with this surface, appear and disappear continuously. Our simulations illuminate the danger of extrapolating low-temperature results to operando conditions and indicate that the catalytic activity can only be inferred from calculations that take dynamics fully into account. More than that, they show that it is the transition to this highly fluctuating interfacial environment that drives the catalytic process.
@article{Bonati2023TheFe111, author = {Bonati, Luigi and Polino, Daniela and Pizzolitto, Cristina and Biasi, Pierdomenico and Eckert, Rene and Reitmeier, Stephan and Schlögl, Robert and Parrinello, Michele}, doi = {10.1073/pnas.231302312}, issn = {10916490}, issue = {50}, journal = {Proceedings of the National Academy of Sciences of the United States of America}, keywords = {enhanced sampling,heterogeneous catalysis,machine learning,molecular dynamics,nitrogen decomposition}, month = dec, pages = {e2313023120}, pmid = {38060558}, publisher = {National Academy of Sciences}, title = {The role of dynamics in heterogeneous catalysis: Surface diffusivity and N2 decomposition on Fe(111)}, volume = {120}, url = {https://pnas.org/doi/10.1073/pnas.231302312}, year = {2023}, }
2022
- Catal. Today
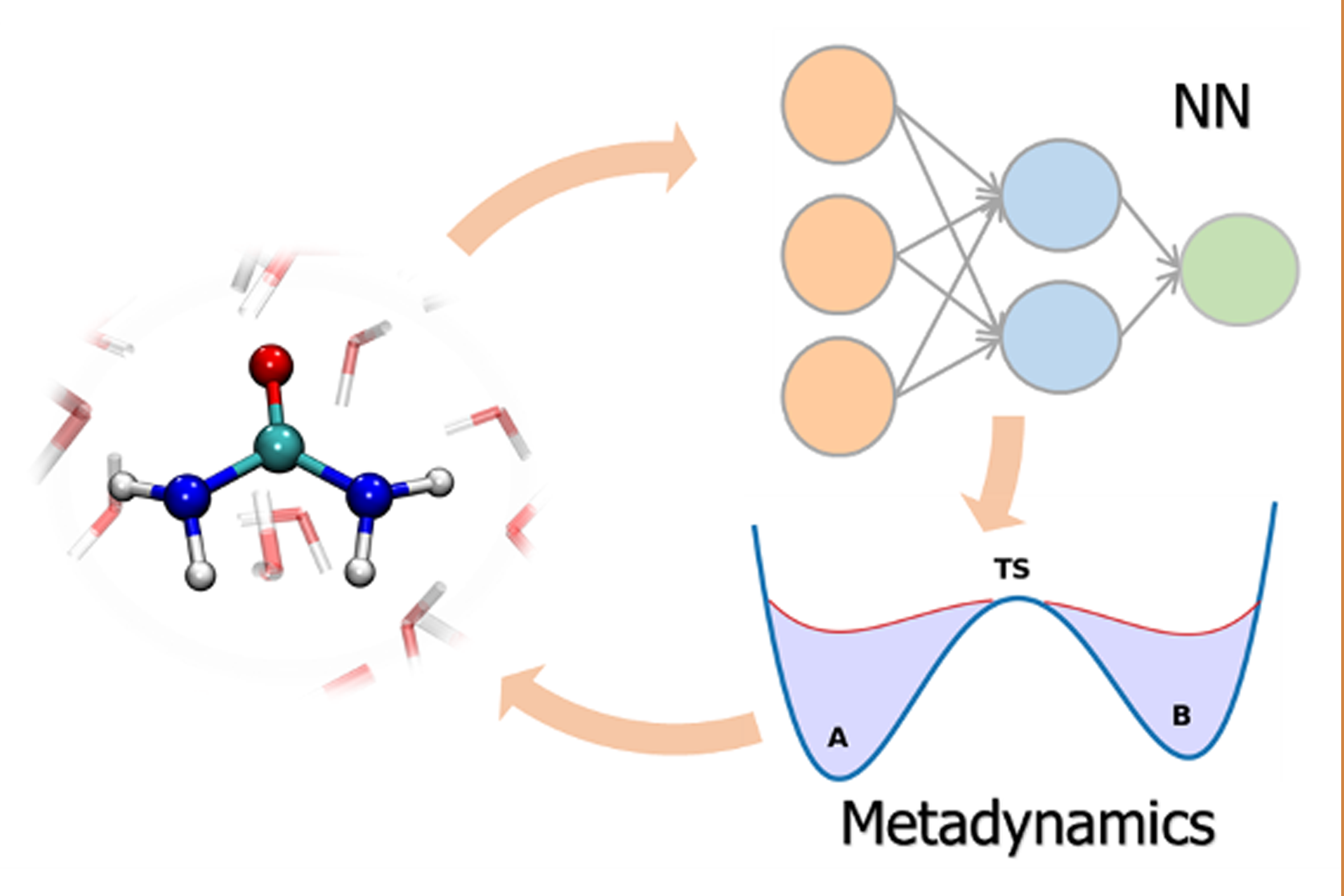 Using metadynamics to build neural network potentials for reactive events: the case of urea decomposition in waterManyi Yang, Luigi Bonati, Daniela Polino, and Michele ParrinelloCatalysis Today, Mar 2022
Using metadynamics to build neural network potentials for reactive events: the case of urea decomposition in waterManyi Yang, Luigi Bonati, Daniela Polino, and Michele ParrinelloCatalysis Today, Mar 2022The study of chemical reactions in aqueous media is very important for its implications in several fields of science, from biology to industrial processes. However, modeling these reactions is difficult when water directly participates in the reaction, since it requires a fully quantum mechanical description of the system. Ab-initio molecular dynamics is the ideal candidate to shed light on these processes. However, its scope is limited by a high computational cost. A popular alternative is to perform molecular dynamics simulations powered by machine learning potentials, trained on an extensive set of quantum mechanical calculations. Doing so reliably for reactive processes is difficult because it requires including very many intermediate and transition state configurations. In this study we used an active learning procedure accelerated by enhanced sampling to harvest such structures and to build a neural-network potential to study the urea decomposition process in water. This allowed us to obtain the free energy profiles of this important reaction in a wide range of temperatures, to discover several novel metastable states, and improve the accuracy of the kinetic rates calculations. Furthermore, we found that the formation of the zwitterionic intermediate has the same probability of occurring via an acidic or a basic pathway, which could be the cause of the insensitivity of reaction rates to the solution pH.
@article{Yang2022UsingWater, author = {Yang, Manyi and Bonati, Luigi and Polino, Daniela and Parrinello, Michele}, doi = {10.1016/J.CATTOD.2021.03.018}, issn = {0920-5861}, journal = {Catalysis Today}, keywords = {Free energy surface,Kinetic rates,Metadynamics,Neural network potentials,Urea decomposition}, month = mar, pages = {143-149}, publisher = {Elsevier}, title = {Using metadynamics to build neural network potentials for reactive events: the case of urea decomposition in water}, volume = {387}, url = {https://www.sciencedirect.com/science/article/pii/S092058612100136X}, year = {2022}, } - JCTC
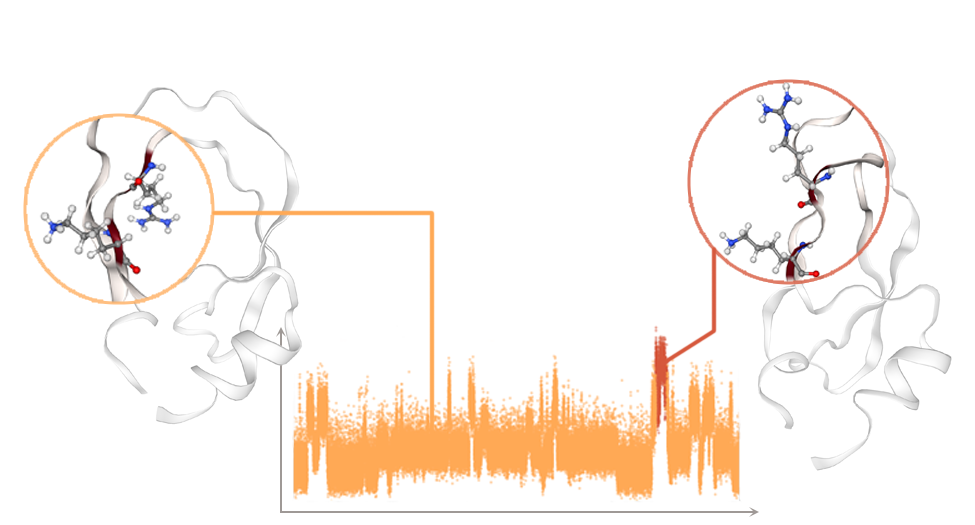 Characterizing Metastable States with the Help of Machine LearningPietro Novelli, Luigi Bonati, Massimiliano Pontil, and Michele ParrinelloJournal of Chemical Theory and Computation, Sep 2022
Characterizing Metastable States with the Help of Machine LearningPietro Novelli, Luigi Bonati, Massimiliano Pontil, and Michele ParrinelloJournal of Chemical Theory and Computation, Sep 2022Present-day atomistic simulations generate long trajectories of ever more complex systems. Analyzing these data, discovering metastable states, and uncovering their nature are becoming increasingly...
@article{Novelli2022CharacterizingLearning, author = {Novelli, Pietro and Bonati, Luigi and Pontil, Massimiliano and Parrinello, Michele}, doi = {10.1021/ACS.JCTC.2C00393}, issn = {15499626}, issue = {9}, journal = {Journal of Chemical Theory and Computation}, month = sep, pages = {5195-5202}, pmid = {35920063}, publisher = {American Chemical Society}, title = {Characterizing Metastable States with the Help of Machine Learning}, volume = {18}, url = {/doi/pdf/10.1021/acs.jctc.2c00393?ref=article_openPDF}, year = {2022}, }
2021
- Nat. Commun.
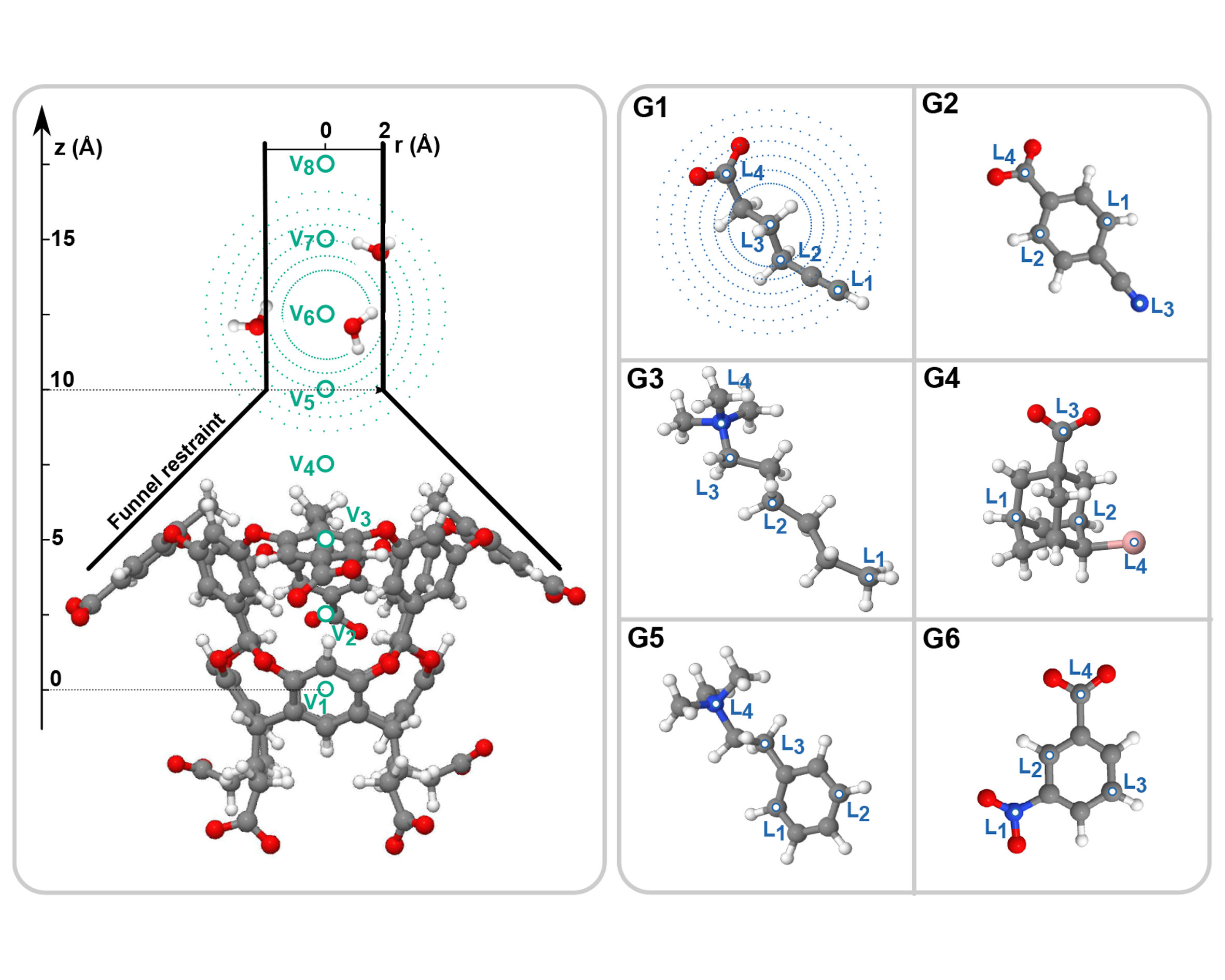 The role of water in host-guest interactionValerio Rizzi, Luigi Bonati, Narjes Ansari, and Michele ParrinelloNature Communications, Sep 2021
The role of water in host-guest interactionValerio Rizzi, Luigi Bonati, Narjes Ansari, and Michele ParrinelloNature Communications, Sep 2021One of the main applications of atomistic computer simulations is the calculation of ligand binding free energies. The accuracy of these calculations depends on the force field quality and on the thoroughness of configuration sampling. Sampling is an obstacle in simulations due to the frequent appearance of kinetic bottlenecks in the free energy landscape. Very often this difficulty is circumvented by enhanced sampling techniques. Typically, these techniques depend on the introduction of appropriate collective variables that are meant to capture the system’s degrees of freedom. In ligand binding, water has long been known to play a key role, but its complex behaviour has proven difficult to fully capture. In this paper we combine machine learning with physical intuition to build a non-local and highly efficient water-describing collective variable. We use it to study a set of host-guest systems from the SAMPL5 challenge. We obtain highly accurate binding free energies and good agreement with experiments. The role of water during the binding process is then analysed in some detail.
@article{Rizzi2021TheInteraction, author = {Rizzi, Valerio and Bonati, Luigi and Ansari, Narjes and Parrinello, Michele}, doi = {10.1038/s41467-020-20310-0}, issn = {20411723}, issue = {1}, journal = {Nature Communications}, pages = {2-8}, publisher = {Springer US}, title = {The role of water in host-guest interaction}, volume = {12}, url = {http://dx.doi.org/10.1038/s41467-020-20310-0}, year = {2021}, } - PNAS
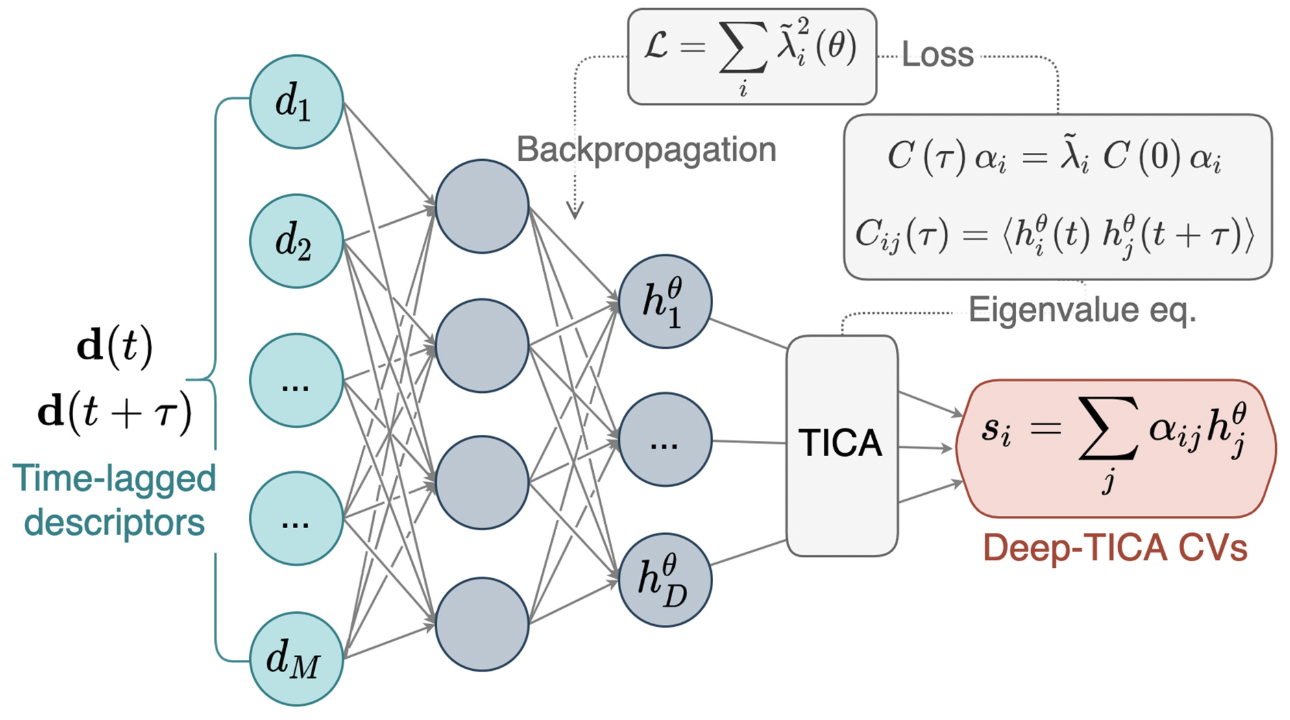 Deep learning the slow modes for rare events samplingLuigi Bonati, GiovanniMaria Piccini, and Michele ParrinelloProceedings of the National Academy of Sciences, 16 2021
Deep learning the slow modes for rare events samplingLuigi Bonati, GiovanniMaria Piccini, and Michele ParrinelloProceedings of the National Academy of Sciences, 16 2021The development of enhanced sampling methods has greatly extended the scope of atomistic simulations, allowing long-time phenomena to be studied with accessible computational resources. Many such methods rely on the identification of an appropriate set of collective variables. These are meant to describe the system’s modes that most slowly approach equilibrium. Once identified, the equilibration of these modes is accelerated by the enhanced sampling method of choice. An attractive way of determining the collective variables is to relate them to the eigenfunctions and eigenvalues of the transfer operator. Unfortunately, this requires knowing the long-term dynamics of the system beforehand, which is generally not available. However, we have recently shown that it is indeed possible to determine efficient collective variables starting from biased simulations. In this paper, we bring the power of machine learning and the efficiency of the recently developed on-the-fly probability enhanced sampling method to bear on this approach. The result is a powerful and robust algorithm that, given an initial enhanced sampling simulation performed with trial collective variables or generalized ensembles, extracts transfer operator eigenfunctions using a neural network ansatz and then accelerates them to promote sampling of rare events. To illustrate the generality of this approach we apply it to several systems, ranging from the conformational transition of a small molecule to the folding of a mini-protein and the study of materials crystallization.
@article{Bonati2021DeepSampling, title = {Deep learning the slow modes for rare events sampling}, author = {Bonati, Luigi and Piccini, GiovanniMaria and Parrinello, Michele}, journal = {Proceedings of the National Academy of Sciences}, doi = {10.1073/pnas.2113533118}, volume = {118}, number = {44}, pages = {e2113533118}, year = {2021}, month = {16}, publisher = {National Academy of Sciences}, } - Nuovo Cimento C
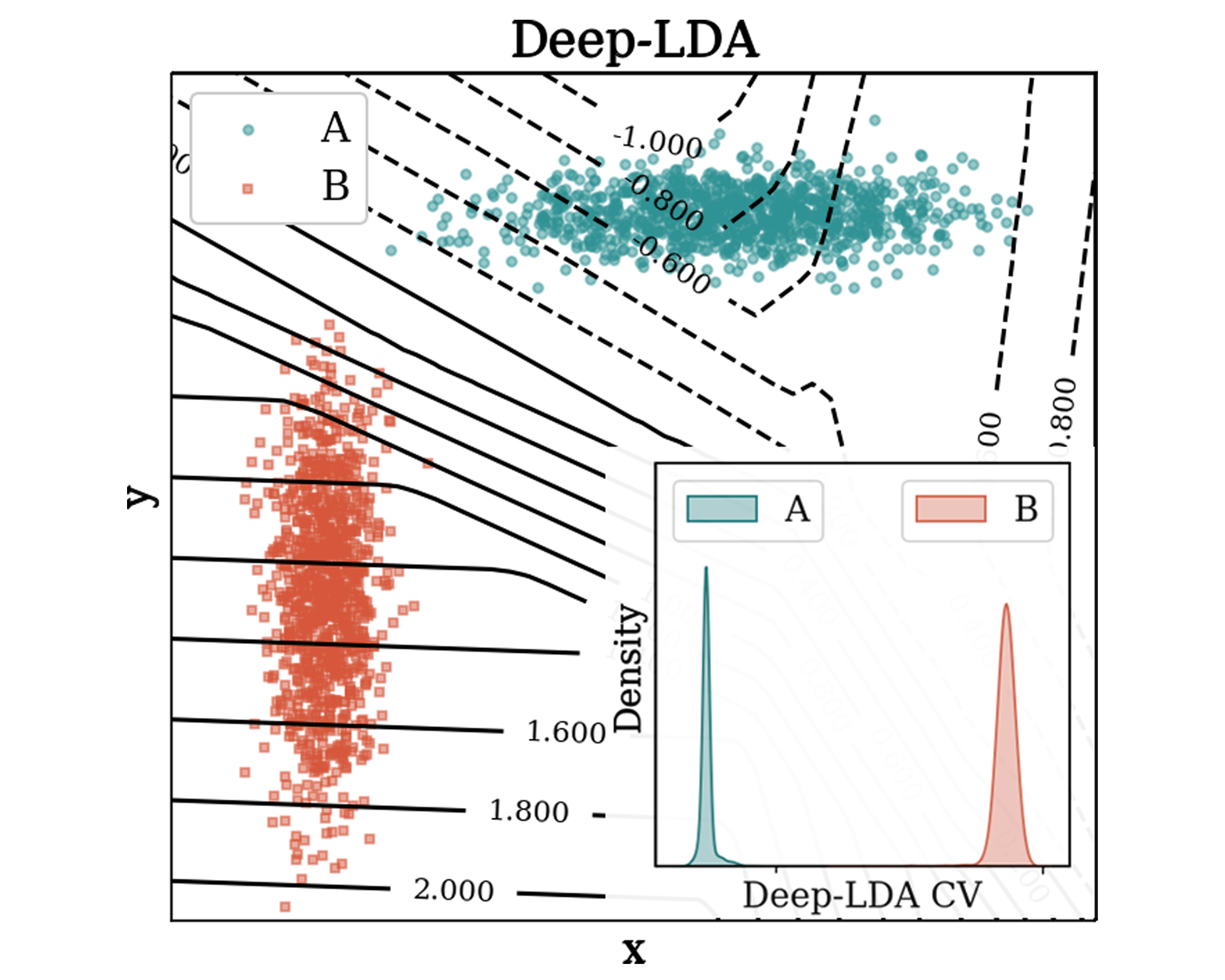 Training collective variables for enhanced sampling via neural networks based discriminant analysisLuigi BonatiNuovo Cimento C, 16 2021
Training collective variables for enhanced sampling via neural networks based discriminant analysisLuigi BonatiNuovo Cimento C, 16 2021A popular way to accelerate the sampling of rare events in molecular dynamics simulations is to introduce a potential that increases the fluctuations of selected collective variables. For this strategy to be successful, it is critical to choose appropriate variables. Here we review some recent developments in the data-driven design of collective variables, with a focus on the combination of Fisher’s discriminant analysis and neural networks. This approach allows to compress the fluctuations of metastable states into a low-dimensional representation. We illustrate through several examples the effectiveness of this method in accelerating the sampling, while also identifying the physical descriptors that undergo the most significant changes in the process.
@article{Bonati2021TrainingAnalysis, author = {Bonati, Luigi}, journal = {Nuovo Cimento C}, title = {Training collective variables for enhanced sampling via neural networks based discriminant analysis}, doi = {10.1393/ncc/i2021-21125-3}, year = {2021}, issue = {4-5}, page = {125}, }
2020
- Nat. Commun.
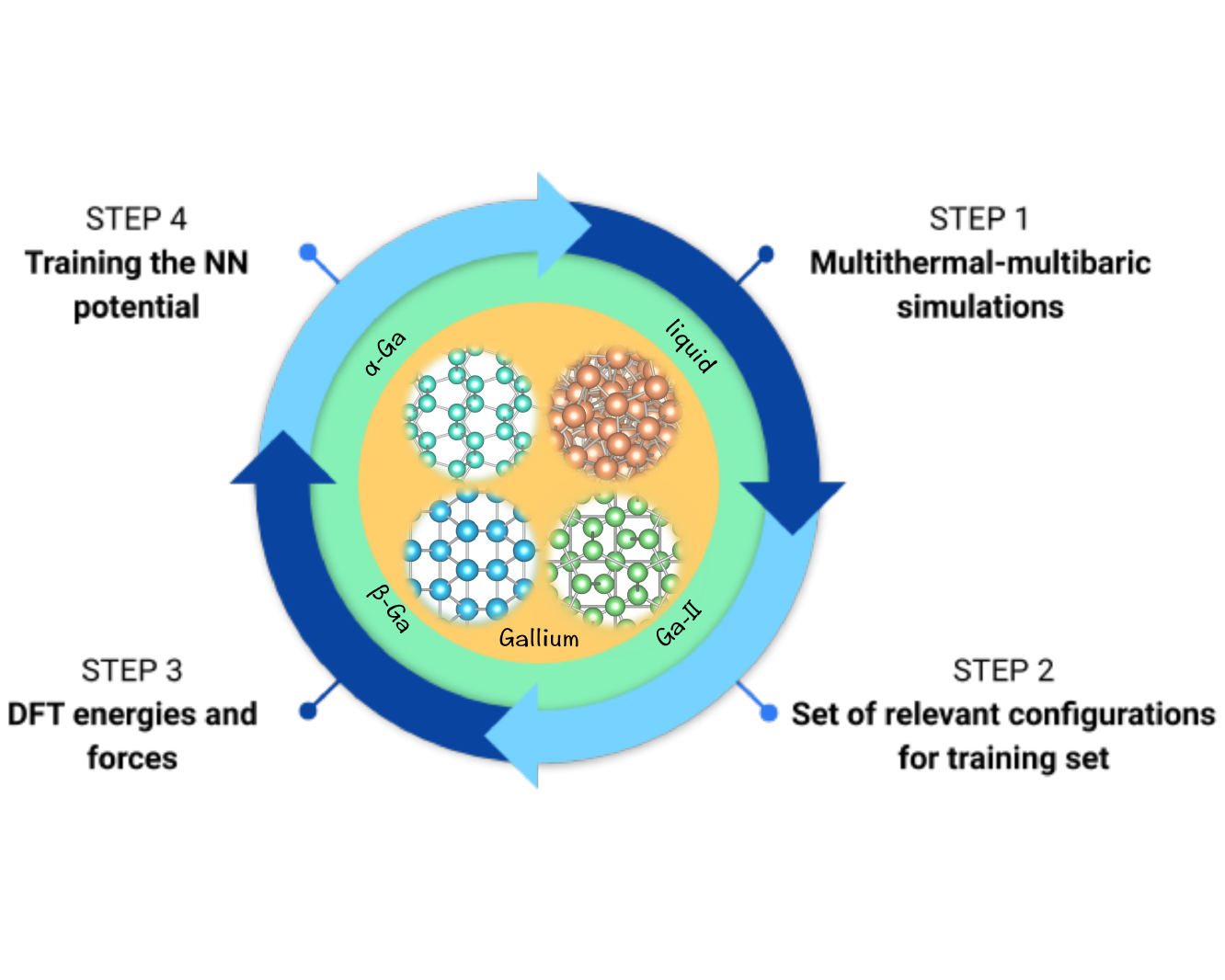 Ab initio phase diagram and nucleation of galliumHaiyang Niu, Luigi Bonati, Pablo M. Piaggi, and Michele ParrinelloNature Communications, 16 2020
Ab initio phase diagram and nucleation of galliumHaiyang Niu, Luigi Bonati, Pablo M. Piaggi, and Michele ParrinelloNature Communications, 16 2020Elemental gallium possesses several intriguing properties, such as a low melting point, a density anomaly and an electronic structure in which covalent and metallic features coexist. In order to simulate this complex system, we construct an ab initio quality interaction potential by training a neural network on a set of density functional theory calculations performed on configurations generated in multithermal–multibaric simulations. Here we show that the relative equilibrium between liquid gallium, α-Ga, β-Ga, and Ga-II is well described. The resulting phase diagram is in agreement with the experimental findings. The local structure of liquid gallium and its nucleation into α-Ga and β-Ga are studied. We find that the formation of metastable β-Ga is kinetically favored over the thermodinamically stable α-Ga. Finally, we provide insight into the experimental observations of extreme undercooling of liquid Ga.
@article{Niu2020AbGallium, author = {Niu, Haiyang and Bonati, Luigi and Piaggi, Pablo M. and Parrinello, Michele}, doi = {10.1038/s41467-020-16372-9}, issn = {20411723}, issue = {1}, journal = {Nature Communications}, pages = {1-9}, pmid = {32461573}, publisher = {Springer US}, title = {Ab initio phase diagram and nucleation of gallium}, volume = {11}, url = {http://dx.doi.org/10.1038/s41467-020-16372-9}, year = {2020}, } - J. Phys. Chem. Lett.
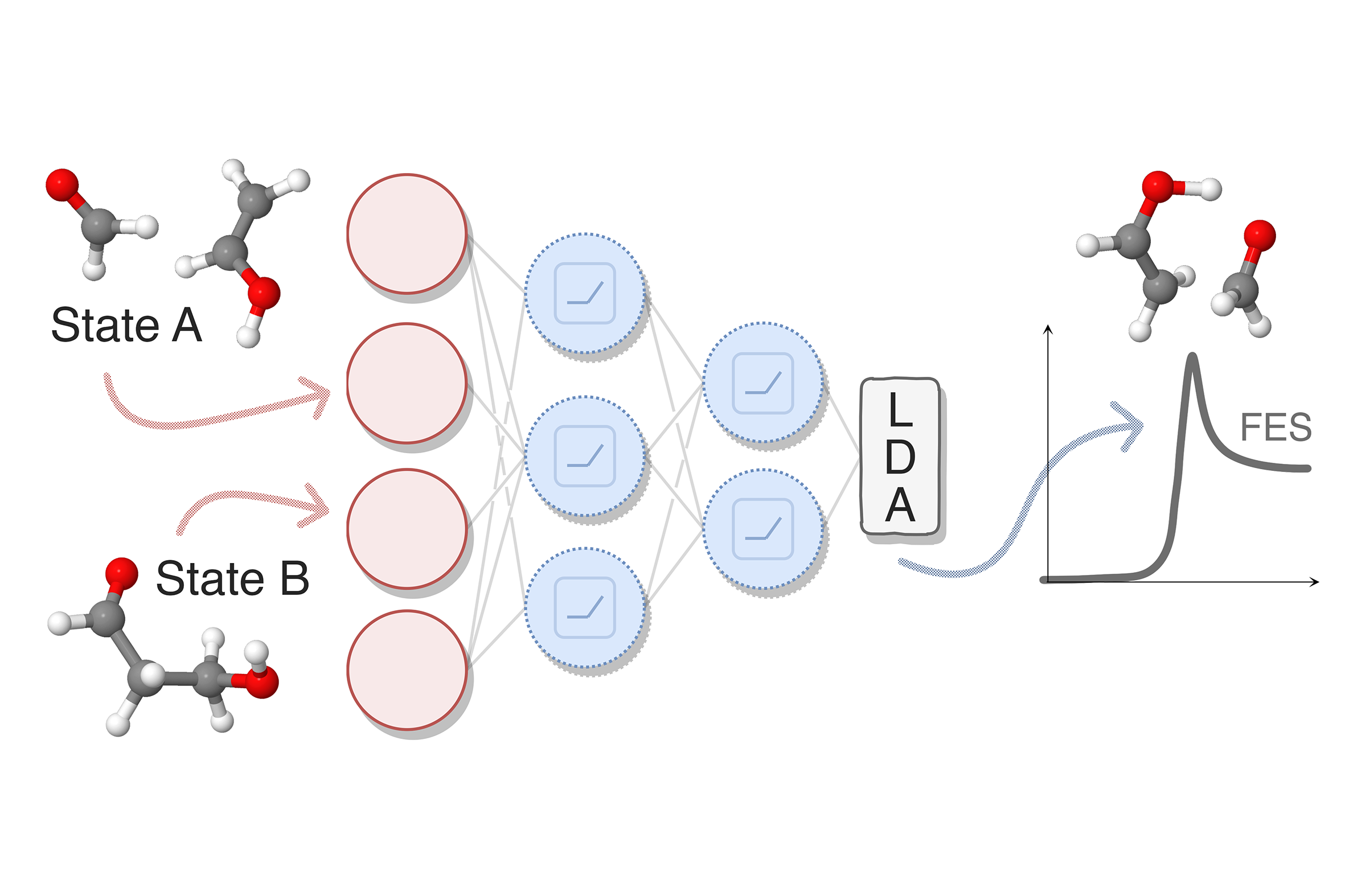 Data-Driven Collective Variables for Enhanced SamplingLuigi Bonati, Valerio Rizzi, and Michele ParrinelloJournal of Physical Chemistry Letters, 16 2020
Data-Driven Collective Variables for Enhanced SamplingLuigi Bonati, Valerio Rizzi, and Michele ParrinelloJournal of Physical Chemistry Letters, 16 2020Designing an appropriate set of collective variables is crucial to the success of several enhanced sampling methods. Here we focus on how to obtain such variables from information limited to the metastable states. We characterize these states by a large set of descriptors and employ neural networks to compress this information in a lower-dimensional space, using Fisher’s linear discriminant as an objective function to maximize the discriminative power of the network. We test this method on alanine dipeptide, using the nonlinearly separable data set composed by atomic distances. We then study an intermolecular aldol reaction characterized by a concerted mechanism. The resulting variables are able to promote sampling by drawing nonlinear paths in the physical space connecting the fluctuations between metastable basins. Lastly, we interpret the behavior of the neural network by studying its relation to the physical variables. Through the identification of its most relevant features, we are able to gain chemical insight into the process.
@article{Bonati2020DataDrivenSampling, author = {Bonati, Luigi and Rizzi, Valerio and Parrinello, Michele}, doi = {10.1021/acs.jpclett.0c00535}, issn = {19487185}, issue = {8}, journal = {Journal of Physical Chemistry Letters}, pages = {2998-3004}, pmid = {32239945}, title = {Data-Driven Collective Variables for Enhanced Sampling}, volume = {11}, url = {https://dx.doi.org/10.1021/acs.jpclett.0c00535}, year = {2020}, }
2019
- PNAS
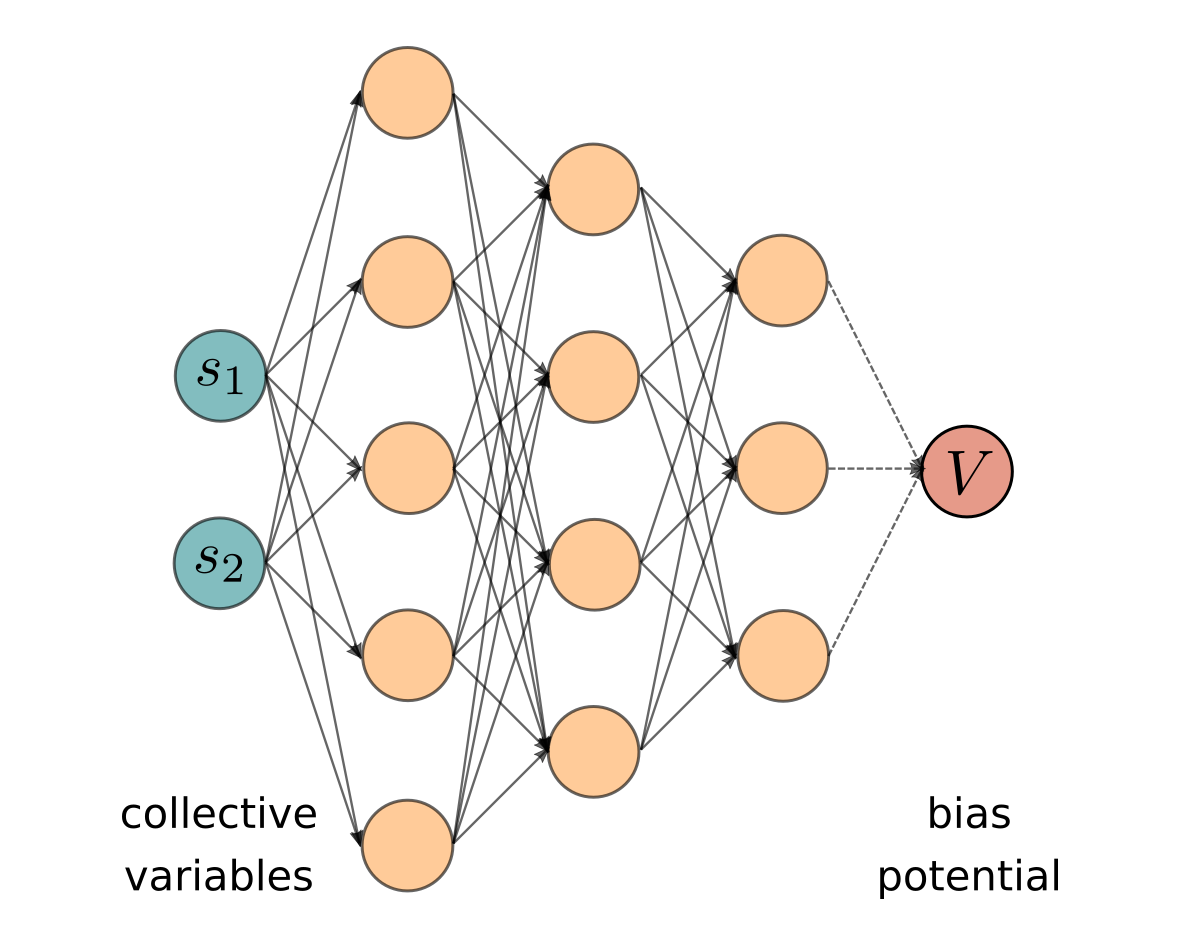 Neural networks-based variationally enhanced samplingLuigi Bonati, Yue Yu Zhang, and Michele ParrinelloProceedings of the National Academy of Sciences of the United States of America, 16 2019
Neural networks-based variationally enhanced samplingLuigi Bonati, Yue Yu Zhang, and Michele ParrinelloProceedings of the National Academy of Sciences of the United States of America, 16 2019Sampling complex free-energy surfaces is one of the main challenges of modern atomistic simulation methods. The presence of kinetic bottlenecks in such surfaces often renders a direct approach useless. A popular strategy is to identify a small number of key collective variables and to introduce a bias potential that is able to favor their fluctuations in order to accelerate sampling. Here, we propose to use machine-learning techniques in conjunction with the recent variationally enhanced sampling method [O. Valsson, M. Parrinello, Phys. Rev. Lett. 113, 090601 (2014)] in order to determine such potential. This is achieved by expressing the bias as a neural network. The parameters are determined in a variational learning scheme aimed at minimizing an appropriate functional. This required the development of a more efficient minimization technique. The expressivity of neural networks allows representing rapidly varying free-energy surfaces, removes boundary effects artifacts, and allows several collective variables to be handled.
@article{Bonati2019NeuralSampling, author = {Bonati, Luigi and Zhang, Yue Yu and Parrinello, Michele}, doi = {10.1073/pnas.1907975116}, issn = {10916490}, issue = {36}, journal = {Proceedings of the National Academy of Sciences of the United States of America}, keywords = {Deep learning,Enhanced sampling,Molecular dynamics}, pages = {17641-17647}, pmid = {31416918}, title = {Neural networks-based variationally enhanced sampling}, volume = {116}, year = {2019}, }
2018
- PRL
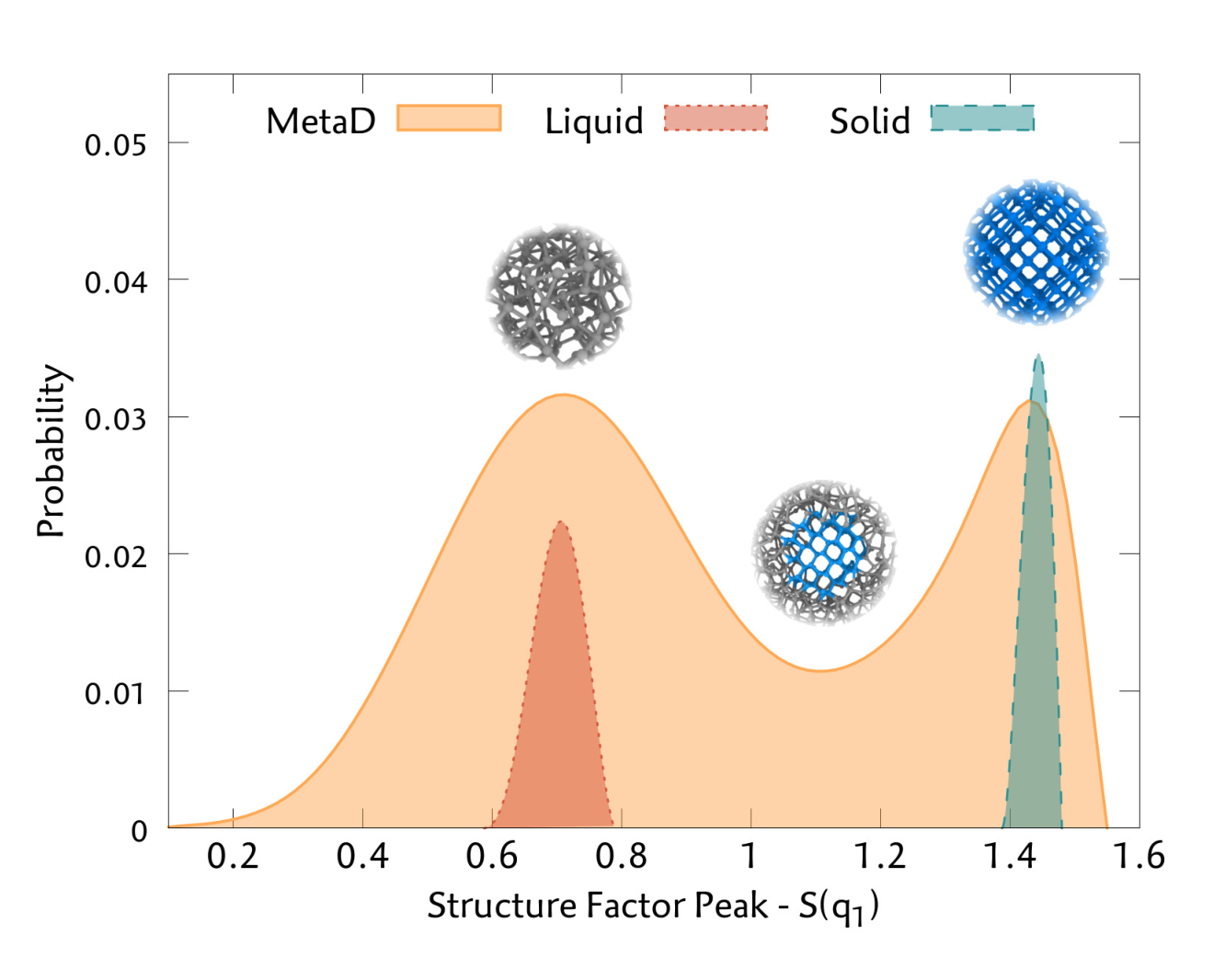 Silicon Liquid Structure and Crystal Nucleation from Ab Initio Deep MetadynamicsLuigi Bonati and Michele ParrinelloPhysical Review Letters, Dec 2018
Silicon Liquid Structure and Crystal Nucleation from Ab Initio Deep MetadynamicsLuigi Bonati and Michele ParrinelloPhysical Review Letters, Dec 2018Studying the crystallization process of silicon is a challenging task since empirical potentials are not able to reproduce well the properties of both a semiconducting solid and metallic liquid. On the other hand, nucleation is a rare event that occurs in much longer timescales than those achievable by ab initio molecular dynamics. To address this problem, we train a deep neural network potential based on a set of data generated by metadynamics simulations using a classical potential. We show how this is an effective way to collect all the relevant data for the process of interest. In order to efficiently drive the crystallization process, we introduce a new collective variable based on the Debye structure factor. We are able to encode the long-range order information in a local variable which is better suited to describe the nucleation dynamics. The reference energies are then calculated using the strongly constrained and appropriately normed (SCAN) exchange-correlation functional, which is able to get a better description of the bonding complexity of the Si phase diagram. Finally, we recover the free energy surface with a density functional theory accuracy, and we compute the thermodynamics properties near the melting point, obtaining a good agreement with experimental data. In addition, we study the early stages of the crystallization process, unveiling features of the nucleation mechanism.
@article{Bonati2018SiliconMetadynamics, author = {Bonati, Luigi and Parrinello, Michele}, doi = {10.1103/PhysRevLett.121.265701}, issn = {0031-9007}, issue = {26}, journal = {Physical Review Letters}, month = dec, pages = {265701}, pmid = {30636123}, title = {Silicon Liquid Structure and Crystal Nucleation from <i>Ab Initio</i> Deep Metadynamics}, volume = {121}, url = {https://link.aps.org/doi/10.1103/PhysRevLett.121.265701}, year = {2018}, }